
 药监信息
药监信息
 现在位置 :
现在位置 :
2010年9月26日国家食品药品监督管理局网站正式发布《2009年药品注册审批年度报告》。报告由2009年药品注册管理工作情况、2009年批准药品生产上市情况、2009年批准药品临床研究情况、2009年重要治疗领域的药品批准情况、2009年药品注册申请受理情况和结语6部分组成。报告共7000余字,图文并茂地展示了2009年药品注册工作的进展与成效。
报告从完善药品注册法规体系、加强药品研究过程的监管、加大技术审评科学公开透明、完成应急防控药品审批四个角度,阐述了2009年药品注册工作的重要举措。
报告显示,2009年的药品注册申请受理总量为6428件,其中,境内申请5128件,包括2336件新申请和2792件补充申请;境外申请1300件,包括新申请614件和补充申请686件。药品注册申请全年受理总量已连续3年稳定在6000至7000件左右。
报告显示,2009年国家局共批准生产上市药品申请3100件(以受理号计,下同),其中含2008年集中审评后续批准的过渡期品种2308件。按照新的《药品注册管理办法》规定,审评并批准了新药、改剂型、仿制药及进口药品注册申请共计792件。其中,化药548件,中药92件,生物制品38件。
2009年化药新药占化药批准品种总数的比例为32%,中药新药占中药批准品种总数的比例为78%。2009年首次出现了批准新药比率升高、重复申请降低的现象。这表明2007年新版《药品注册管理办法》颁布以后,我国采取的一系列规范审评、鼓励创新的政策导向产生了良好效应。
通过开展药品注册现场核查、药品批准文号清查、过渡期品种集中审评、实施新修订的《药品注册管理办法》及其配套法规文件等措施,经过连续几年的努力,药品审评审批改革已经取得显著成效,药品注册管理不断规范,药品研制秩序持续好转,药品注册申请数量趋于理性,质量进一步提高,有效保障了人民群众用药安全有效。
从今年起,国家食品药品监督管理局将逐步建立和完善药品注册审批年度报告制度,积极推进药品注册审批信息公开和工作透明,为鼓励新药创制,促进医药产业结构调整和产品结构优化提供有效指导。
2009年药品注册审批年度报告
国家食品药品监督管理局
2010年8月
目 录
1. 2009年药品注册管理工作情况
2. 2009年批准药品生产上市情况
3. 2009年批准药品临床研究情况
4. 2009年重要治疗领域的药品批准情况
5. 2009年药品注册申请受理情况
6. 结语
药品注册,是国家食品药品监督管理局依照《药品管理法》的规定,根据药品注册申请人的申请,对拟上市销售药品的安全性、有效性、质量可控性等进行审查,并决定是否同意其申请的审批过程。其根本目的是通过科学评价保证上市药品安全有效,保障和促进公众健康。在药品研制、生产、流通、使用的全过程监管中,药品注册管理是从源头上对药品安全性和有效性实施监管的重要手段。
1. 2009年药品注册管理的重要举措
2009年,药品注册管理工作深入贯彻落实科学发展观,大力实践科学监管理念,以风险效益评估和风险管理为核心,坚持“规范审批、公开透明、鼓励创新”的原则,完善药品注册法规,健全药品审评审批机制,提高审评审批效率,较好地履行了《药品管理法》赋予的职责。
1.1 完善药品注册法规体系
一是加快制定《药品注册管理办法》配套文件。颁布了《新药注册特殊审批管理规定》对于符合特殊审批条件的注册申请,实行“早期介入、优先审评、多渠道沟通交流、动态补充资料”,充分体现了鼓励创新和加强风险控制管理的导向,促进我国创新药的研究与开发。出台了《药品技术转让注册管理规定》,促进产品结构调整和生产资源的合理分配,实现优势产品重组和重点发展。出台了《药物临床试验机构资格认定复核检查标准》,开展临床试验复核检查,加强对临床研究机构的监管。完成《药品标准管理办法》、《药用原辅材料备案管理规定》、《天然药物注册管理补充规定》、《SAE报告和管理规定》和《伦理委员会技术指导原则》的初步起草工作,进一步加快药品注册管理法规体系的完善。
二是健全药物研究技术指导原则体系。采取先翻译后转化的办法,借鉴和引入国际公认并遵循的药物研发指南,有计划、分步骤地推进我国指导原则体系建立。制定了复方抗生素研究技术要求、银杏叶和丹参类中药注射剂安全性及上市后风险评估技术要求、《药物致癌试验必要性的技术指导原则》等,着力解决药品安全问题。开展了《通用技术文件(CTD)格式申报资料提交要求》研究,逐步规范药品研发和技术审评。
1.2 加强药品研究过程的监督管理
一是规范和加强药品注册现场核查。各省局严格按照《药品注册现场核查管理规定》的要求,结合实际情况,制定了本省药品注册现场核查工作实施细则、工作程序及核查人员工作守则和管理规定,使药品注册现场核查工作有序、规范、廉洁地开展,较好地完成了药品注册现场核查工作。2009年,全国共完成3721个注册申请的注册现场核查,较好地保证了药品申报资料的真实性。
二是开展GCP复核检查。在试点检查的基础上,制定了GCP复核检查工作程序、检查标准,分区域逐步实施了复核检查。按照药物研究监督与注册审评相结合、机构监督检查和品种检查相结合的工作要求,以风险管理为基础,以研究品种抽查为重点的检查原则,对临床研究机构执行GCP情况开展了评估。
1.3 确保技术审评科学公开透明
一是进一步提高审评效率。采取科学分类审评任务、创新优化工作模式、合理调配审评资源、加强工作调度与管理、加大审评计划的制定与监督等措施,逐步实现了按时限审评。
二是进一步加大技术审评公开透明力度。加强与药品注册申请人的沟通与交流,使其准确把握和理解技术审评要求和审评关键问题,保证审评的科学性,提高技术审评的效率和工作透明度。在甲型H1N1流感疫苗审评中,首次采用了公开审评的方式,对10家企业的疫苗进行现场表决,收效良好。2009年,在网站公开了70例审评案例;举办研讨班14期,4000余人次参加;举办开放日活动11次,接待281人次;召开专家咨询会11次,沟通交流会80次,全年共接待4000余人次咨询。
三是进一步提高技术审评工作的科学性和公正性。通过制定注册申报资料受理审查要点,开展第三方验证,利用药品注册研制现场核查、药品生产现场检查等技术监督措施,全面支撑和保障药品注册审评工作顺利开展,提升了申报资料的真实性和可追溯性。
1.4 完成应急防控药品审批
为应对甲型H1N1流感所致的突发公共卫生事件,根据国家防控工作的整体要求,依据《国家食品药品监督管理局药品特别审批程序》,及时制定了《大流行流感疫苗特别审批应急工作方案》、《甲型H1N1流感疫苗审评审批工作方案》、《甲型H1N1流感疫苗研发技术考虑要点》等应急防控的配套指南。采取早期介入、全面协调、关口前移、现场核查等一系列措施,在保证安全、有效的前提下,启动了“特别审批程序”,完成了甲型H1N1流感疫苗的审批,为疫情防控提供了有效手段,也为应急防控药物审批积累了宝贵的经验。
2. 2009年药品批准生产上市情况
2.1 批准生产上市情况
2009年,批准生产上市药品申请3100件,其中含2008年集中审评后续批准的过渡期品种2308件。按照新的《药品注册管理办法》规定,审评并批准了新药、改剂型、仿制药及进口药品注册申请共计792件,其中,化药批准上市药品548件,占80%;中药92件,占14%,且与以往相比批准数量明显降低;生物制品38件,占6%。批准境内生产药品678件,占全年批准生产上市药品的86%,进口药品114件,占14%。见表1。
表1 2009年批准生产上市的药品
|
注册类型 |
批准境内生产 |
批准进口上市 | |||
|
新药 |
改剂型 |
仿制药 |
小计 | ||
|
化学药品 |
175 |
17 |
356 |
548 |
100 |
|
中 药 |
72 |
8 |
12 |
92 |
1 |
|
生物制品 |
38 |
38 |
13 | ||
|
合计 |
678 |
114 | |||
|
总计 |
792 | ||||
注:①表中数据以受理号计,受理号系申请人提出的一件申请事项的编号。各申请企业的原料药、制剂、制剂不同规格分别予以编号。
②表中新药系根据《药品注册管理办法》规定按照新药管理的药品。化药新药包括化学药品注册分类1-4,中药新药包括中药、天然药物注册分类1-7,生物制品注册分类1-13类;
③表中化药改剂型为化学药品注册分类5,中药改剂型为中药、天然药物注册分类8,生物制品改剂型为生物制品注册分类14;
④表中化药仿制药为化学药品注册分类6,中药仿制药为中药、天然药物注册分类9,生物制品仿制药为生物制品注册分类15。
2.2 批准品种的结构情况
新药与仿制药的比率是反映当前药品研发走势与注册申报结构的一个客观指标,同时也反映了药品的审评审批工作情况。数据表明,2009年化药新药占化药批准品种总数的比例为32%(以受理号计),中药新药占中药批准品种总数的比例为78%。2009年首次出现了批准新药比率升高、重复申请降低的现象。这表明2007年新版《药品注册管理办法》颁布以后,我国采取的一系列规范审评、鼓励创新的政策导向产生了良好效应。
由于原料药以及不同的制剂规格将分别给予受理号,不同厂家同一品种也分别给予受理号,所以相同化合物或中药处方可能有多个受理号。化合物或中药处方与其受理号的比例,在一定程度上可以反映同一品种重复申报的情况。2009年化药新药化合物与受理号的比例为1:1.9;仿制药为1:2.5,均较去年明显下降(2008年化药新药1:2.5,仿制药1:3)。2009年批准生产药品的受理号与化合物(或处方)之间的关系见表2。
表2 2009年批准生产药品的受理号与化合物(或处方)之间的关系
|
|
化药 |
中药 | ||||
|
新药 |
改剂型 |
仿制药 |
新药 |
改剂型 |
仿制药 | |
|
受理号 |
175 |
17 |
356 |
72 |
8 |
12 |
|
化合物 (或处方) |
94 |
16 |
142 |
65 |
8 |
11 |
|
比值 |
1.9:1 |
1.1:1 |
2.5:1 |
1.1:1 |
1:1 |
1:1 |
2.3 批准1类新药情况
2009年共批准了1类新药12件,其中化学药品10件,生物制品2件。自2006年国务院开展全国整顿和规范药品市场秩序专项行动以来,国家食品药品监督管理局通过开展药品注册现场核查、药品批准文号清查、过渡期品种集中审评、实施新修订的《药品注册管理办法》及其配套法规文件等措施,为我国药物研发由以仿为主逐步走向仿创结合的道路奠定了良好基础。2009年批准1类新药情况见表3。
表3 2009年批准的1类新药
|
药品名称 |
注册类别 |
剂 型 |
申请企业 |
|
盐酸安妥沙星 |
化药1.1类 |
原料药 |
安徽环球药业股份有限公司 |
|
盐酸安妥沙星片 |
化药1.1类 |
片剂 |
安徽环球药业股份有限公司 |
|
左奥硝唑 |
化药1.3类 |
原料药 |
南京圣和药业有限公司 |
|
左奥硝唑氯化钠注射液 |
化药1.3类 |
注射剂 |
南京圣和药业有限公司 |
|
利福平异烟肼片 |
化药1.5类 |
片 剂 |
沈阳红旗制药有限公司 |
|
尼群洛尔片 |
化药1.5类 |
片 剂 |
江苏吉贝尔药业有限公司 |
|
乙胺吡嗪利福异烟片 |
化药1.5类 |
片 剂 |
沈阳红旗制药有限公司 |
|
注射用头孢曲松钠他唑巴坦钠 |
化药1.5类 |
注射剂( |
海口奇力制药有限公司 |
|
注射用头孢曲松钠他唑巴坦钠 |
化药1.5类 |
注射剂( |
海口奇力制药有限公司 |
|
注射用头孢噻肟钠舒巴坦钠 |
化药1.5类 |
注射剂 |
湘北威尔曼制药有限公司 |
|
冻干A、C群脑膜炎球菌多糖结合疫苗 |
预防用生物制品1类 |
注射剂 |
玉溪沃森生物技术有限公司 |
|
口服重组幽门螺杆菌疫苗 |
预防用生物制品1类 |
注射剂 |
重庆康卫生物科技有限公司, 中国人民解放军第三军医大学 |
3. 2009年批准药品临床研究情况
3.1 临床实验批准情况
2009年依据《新药注册特殊审批管理规定》,对符合要求的28个临床试验申请按照特殊审批程序进行了审评审批,并在关键阶段与申请人进行了交流、沟通与讨论,有力地指导和促进了我国新药的研发。全年共计批准1105个药品进入临床试验,其中境内申请785个,境外申请320个。批准了13个(1.1类)化药新化合物、1个(1类)中药新有效单体和48个中药新处方(6类)进入临床试验。
批准进入临床试验药物的适应症既涵盖在我国疾病谱中占重要位置的常见疾病和多发疾病,如肿瘤、心血管病等,也包括了社会影响度高的一些罕见性疾病,如法布雷氏病。具体构成见表4。
表4 2009年批准进入临床试验的药物
|
|
境内申请 |
境外申请 | ||
|
化药 |
中药 |
生物制品 | ||
|
受理号 |
658 |
101 |
26 |
320 |
|
化合物 (或处方) |
267 |
91 |
/ |
171 |
注:化药含人体生物等效性试验
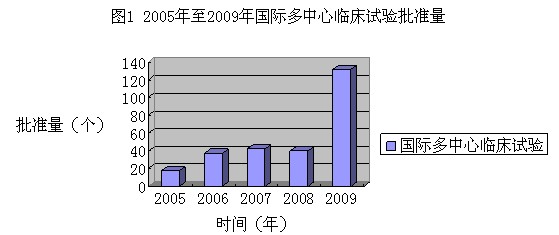
3.2 国际多中心临床试验批准情况
我国参与全球新药研发同步研究的程度逐年加大。2009年批准的320件境外申请人的临床试验中,有132件为国际多中心临床试验申请,较往年明显增加。近5年国际多中心临床试验申请批准情况见图1。
4. 2009年重要治疗领域的药品批准情况
2009年批准的药品主要集中在以下重要治疗领域: 4.1 防治甲型H1N1流感药品 按照“特别审批程序”批准了国内10家疫苗生产企业研发的甲型H1N1流感病毒裂解疫苗。在疫情肆虐期,全球甲流药品短缺的情况下,批准了国产磷酸奥司他韦扩大生产规模、缩短生产工艺的补充申请。批准了磷酸奥司他韦扩大用药人群的补充申请,为特殊人群(儿童、婴幼儿)提供了甲流防控的手段。批准了扎那米韦吸入性粉雾剂的进口申请。上述疫苗和药品的上市,为甲流防控提供了有效保障。 4.2 治疗HIV感染药品 目前,用于治疗HIV感染患者的鸡尾酒疗法的主要药品已在我国批准上市,其中大部分药品已经国产。根据WHO推荐的最新版“艾滋病治疗指南”,以及我国“中国艾滋病病人抗病毒治疗研究”的成果,批准了国产奈韦拉平、齐多夫定、拉米夫定联合用药的研究申请。这对阻断艾滋病的母婴传播以及临床治疗耐药具有重要意义。 批准了拉替拉韦钾片与其他抗逆转录病毒药物联合用于对多种抗逆转录病毒药物耐药的患者,这是第一个在我国批准上市的HIV-1整合酶抑制剂。 批准了非核苷类逆转录酶抑制剂依曲韦林片用于治疗以前曾接受过抗逆转录病毒药物治疗的HIV-1感染成年患者(包括对非核苷类逆转录酶抑制剂耐药的患者)。 上述药品为耐药的HIV感染患者提供了新的治疗手段。 4.3 治疗肿瘤的药品 批准了国内生产企业研发的非洛他赛进入临床试验,这可能对多西他赛耐药的患者提供新的治疗手段;还批准了酪氨酸激酶抑制剂法米替尼进入临床试验。 批准了具有规模化红豆杉种植基地支撑的紫杉醇的生产。批准尼洛替尼进口,为对伊马替尼耐药或不能耐受的慢性髓性白血病患者提供了新的治疗手段;批准了多靶点酪氨酸激酶抑制剂舒尼替尼用于治疗晚期、不可手术的肾癌。 4.4 治疗乙肝的药品 批准了境内抗乙型肝炎治疗药品恩替卡韦的生产。国产药品的获准上市,降低了患者的治疗费用,增加了乙肝治疗药品的可获得性。 4.5 治疗心血管疾病的药品 批准了左西孟旦仿制药的生产。该药品系钙增敏剂类正性肌力药品,用于失代偿性心功能不全患者。批准了国内企业研发的依普立酮的临床试验,该药是目前发现的首个高选择性盐皮质激素受体阻断剂,对高血压和心肌梗塞后心衰患者的治疗具有重要意义。 批准了全新作用机制的高血压治疗药品阿利吉仑进口。该药品为高效选择性人类肾素抑制剂,为高血压的治疗提供了新的手段。 4.6 治疗糖尿病的药品 批准了磷酸西格列汀片和艾塞那肽注射液的进口申请。前者属高选择性DPP-Ⅳ抑制剂,目前为糖尿病的一线治疗药物。后者为肠促胰岛素分泌激素类似物,有与肠促胰岛素分泌激素类似的增强葡萄糖依赖性胰岛素分泌和其他抗高血糖作用,为口服降糖药疗效不佳的Ⅱ型糖尿病患者提供了新的治疗手段。 4.7 抗感染药品 批准了达托霉素的进口。该药品为万古霉素耐药患者提供了临床治疗选择。批准我国首创的具有自主知识产权的全新药品左旋奥硝唑和盐酸安妥沙星生产。 4.8 抗排斥免疫抑制药品 批准了咪唑立宾片仿制药的生产。该药品用于预防肾移植后的器官排斥反应。该品种的国产化大大降低了肾移植患者临床治疗费用,提高患者用药的可获得性。 4.9 抗疟疾药品 批准了国产的青蒿琥酯盐酸阿莫地喹双层片上市,为WHO国际药品采购提供了保障。 4.10 中药复方制剂 批准的中药新复方制剂涉及骨伤科、呼吸科、五官科、妇科等多个治疗领域,为这些治疗领域提供了新品种。 5.2009年药品注册申请受理情况 5.1 注册申请受理总体情况 2009年药品注册申请受理总量为6428件,其中,境内申请5128件,包括2336件新申请和2792件补充申请;境外申请1300件,包括新申请614件和补充申请686件。见表5。 表5 2009年药品注册申请受理情况 注册类型 境内申请 境外申请 小计 新注册申请 补充申请 新注册申请 补充申请 化药 2010 2210 513 569 5302 中药 222 482 3 19 726 生物制品 104 100 98 98 400 合计 2336 2792 614 686 5128 1300 6428 注:以上数据以受理号计。 2009年药品注册申请受理情况与历年受理情况相比,近三年来受理量保持平稳,见表6和图2。 表6 2005年至2009年药品注册申请受理量 境内新注册申请 境外新注册申请 补充申请 合计 2009年 2336 614 3478 6428 2008年 2634 593 3235 6462 2007年 3245 603 3226 7075 2006年 16728 448 4187 21363 2005年 22166 518 3710 26394 注:以上数据以受理号计;补充申请包括境内、境外的补充申请。 5.2 药品注册申请受理变化趋势 一是申报数量保持平稳。2009年共受理了药品注册申请6428件(以受理号计),药品注册申请全年受理总量已连续3年稳定在6000至7000件左右。其主要原因是境内企业的申请数量大幅下降,2009年境内企业申报了2336件药品注册申请,与2005年2万多件申报量相比,已回归正常,保持平稳状态。 二是重复申报明显减少。化合物或中药处方与受理号的比值可以反映化药或中药品种重复申报情况。2005年、2006年这一比值约为1:5。2009年度数据显示,化药这一比值仅为1:1.9至1:2.7,中药几乎没有重复申报品种,见表7。数据表明低水平重复申报已经得到了扭转。 表7 2009年申请药品的受理号与化合物(或处方)的关系 化药 中药 新药 改剂型 仿制药 新药 改剂型 仿制药 受理号 715 191 1104 114 57 51 化合物 或处方 283 126 402 109 45 45 比值 2.5:1 1.5:1 2.7:1 1:1 1.3:1 1:1 三是申报结构保持合理。境内申请中,化药新药、改剂型、仿制药申请,分别占化药总注册申报量的33%、10%、55%,见图3。中药新药、改剂型、仿制药申请,分别占中药总注册申报量的51%、26%、23%,见图4。生物制品继续保持往年申报态势,共计104件新申请。
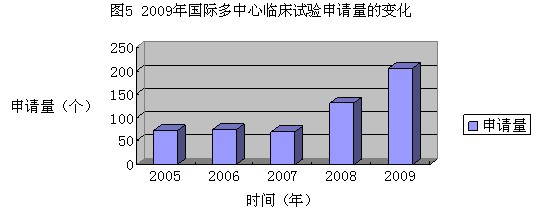
四是国际多中心临床试验申请呈现逐年递增趋势。境外新申请中有205件国际多中心临床试验申请,近两年此类申请呈增加趋势,见图5。 五是研发规范化程度有所改进。申报资料的规范化程度较以往有很大提高,以往不注重生物利用度/生物等效方法学研究问题有所下降,质控方法专属性、注射剂无菌保证研究有所提高。企业更加注重按照药品研发指导原则的要求,客观、规范、严谨地考查药品的安全性和有效性。在新的市场环境下,制药企业更加注重调整产品结构,更加注重产品创新,逐步向以质量求生存、靠创新求发展的模式转变。 6.结语 经过连续几年的努力,药品审评审批改革已经取得显著成效,药品注册管理不断规范,药品研制秩序持续好转,药品注册申请数量趋于理性,质量进一步提高,有效保障了人民群众用药安全有效,为鼓励创制新药和调整医药产业结构奠定了良好基础。 展望2010年,药品注册管理工作要以贯彻落实《药品注册管理办法》及其配套规定为重点,突出抓好“新、优、同、实”核心理念的落实,严把产品准入门槛,严格审评审批标准;进一步加快推进药品标准提高行动计划的执行,重点做好基本药物、民族药、中药注射剂及多组分生化药质量标准提高,全面提升药品质量控制水平,不断提升我国上市药品的安全、有效和质量可控性,为促进公众健康做出更大的贡献! (来源:SFDA网站 2010-9-26)

 京公网安备 11010802023571号
京公网安备 11010802023571号