Law & Regulations
Law & Regulations
 Position :
Position :
Center for Drug Evaluation, NMPA
June 2024
I. Introduction
In order to guide vaccine marketing authorization holders (hereinafter referred to as MAHs) to carry out post-marketing studies of pharmaceutical changes of vaccines, guide and promote the continuous improvement of vaccine manufacturing process, strengthen the supervision and administration of pharmaceutical changes of marketed vaccines and ensure the safety, effectiveness and quality controllability of vaccines, the Guidelines were formulated in accordance with the provisions and requirements of the Drug Administration Law of the People's Republic of China, Vaccine Administration Law of the People's Republic of China, Provisions for Drug Registration, Provisions for the Supervision and Administration of Drug Production, and Provisions for the Administration of Post-marketing Changes of Drugs (Interim).
For the purpose of the Guidelines, vaccines refer to preventive biological products intended for human immunization for the purpose of preventing and controlling the occurrence and epidemic of diseases, covering the pharmaceutical changes of common vaccines marketed in China, such as inactivated vaccines, attenuated live vaccines, subunit vaccines, genetic engineering vaccines, conjugate vaccines and combined vaccines. For vaccines manufactured with new technologies (e.g., mRNA vaccines), the study on changes to the manufacturing process can be carried out by referring to other relevant guidelines at home and abroad and the basic concept of these Guidelines.
Since the pharmaceutical changes of vaccines after marketing are complex and diverse, even if the changes are the same, the risks for different varieties are different. Therefore, when using the Guidelines, the MAH shall implement changes based on adequate risk assessment and change study in combination with the specific vaccine changes. The requirements for the specific study work can be found in the promulgated technical guidelines for vaccines and biological products.
Vaccines are used in healthy populations and involve major public health issues. The MAH is recommended to make adequate assessment and planning for post-marketing changes in advance to minimize the unexpected risks caused by the changes.
The Guidelines were drafted on the basis of the Technical Guidelines for Studies of Pharmaceutical Changes of Marketed Biological Products (Interim) for the particularities and characteristics of vaccines and focusing on the characteristics in vaccine changes (especially changes in the manufacturing process). The Guidelines no longer simply repeat common issues, such as the changes in specifications already included in the Technical Guidelines for Studies of Pharmaceutical Changes of Marketed Biological Products (Interim). For changes within the scope of these Guidelines, these Guidelines shall prevail; for other pharmaceutical changes beyond the scope of the Guidelines, please refer to the Technical Guidelines for Studies of Pharmaceutical Changes of Marketed Biological Products (Interim).
These Guidelines only reflect the current scientific understanding of vaccines and will continue to be improved and updated as scientific research progresses. When applying these Guidelines, reference shall also be made to guidelines such as those of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH).
II. Basic Considerations
MAHs are the main bodies responsible for the post-marketing change management of vaccines, assuming the obligation of managing the whole life cycle of vaccines to ensure that vaccines meet the continuously updated technical requirements after marketing. MAHs shall strengthen the construction of vaccine quality system, be responsible for all post-marketing pharmaceutical change studies, self-assessment of study results and continuous and dynamic change management, and continue to optimize the manufacturing process to maintain the advancement of production and control. See the Technical Guidelines for Studies of Pharmaceutical Changes of Marketed Biological Products (Interim) for the basic considerations regarding the subject responsibility and continuous compliance of post-marketing pharmaceutical changes of vaccines, risk assessment and management of changes, comparability study of changes, associated changes, and changes to excipients and packaging materials. The basic considerations are the basis and principles for the success of vaccine changes and shall be fully implemented in the study of vaccine changes. In addition, based on the characteristics of vaccines, particular attention shall be paid to the following basic considerations:
1. Associated Changes
Post-marketing changes to vaccines are often not independent, and one change may accompany or trigger other changes, which are called associated changes. Due to the complexity of the vaccine system and possible interactions between components, the associated changes and their cumulative risks shall be paid attention to during vaccine changes, and the potential impact of the proposed changes on subsequent steps and related process control parameters shall be carefully considered. For example, the change in production site may be accompanied by the change in the production equipment and manufacturing process. The change in vaccine diluent (especially diluent containing antigen component or adjuvant component) and the change in a single component of a combined vaccine may affect other components and the formulation system, and may affect the overall safety, efficacy and quality controllability of the vaccine.
For associated changes, the study shall be conducted separately with reference to the requirements of each change, and the overall change comparability study shall be conducted, which shall be classified according to the highest change category. When necessary, adequate consideration shall be given to collecting data from different stages of key intermediates, drug substance, and drug product to support the conclusion of comparability and to demonstrate the combined impact on the safety, efficacy, and quality controllability of the drug product.
2. Adjuvants
Since there are multiple combinations of different adjuvants and antigens in the marketed vaccine drug products, and the overall drug product study and clinical validation of adjuvants and antigens are required, the study data used to support changes to adjuvants and the adjuvant system shall be specified according to the specific situation of the product characteristics, clinical validation data and degree of change. Technical guidelines for adjuvants other than aluminium adjuvants will be specified separately.
3. Study on Quality Characteristics
When implementing a change, the strategy and scope of the comparability study shall be determined based on the item and type of the change, the anticipated impact of the change on the product, and an assessment of the potential impact of the change on product safety and efficacy.
Major changes to vaccines often require comprehensive post-marketing quality change studies. Comprehensive quality comparability study of post-marketing changes of vaccines mainly includes comparability of manufacturing process and process control, comparability of release test and extended characterization study, comparability of stability, comparability of animal efficacy and safety, etc. A comprehensive analysis in combination with the above aspects shall be conducted. For details, see the Technical Guidelines for Studies of Pharmaceutical Changes of Marketed Biological Products (Interim).
For extended characterization studies of different types of vaccine antigens and drug products, please refer to the Technical Guidelines for Quality Comparability Study of Changes in Vaccine Production Site. The studies shall cover physicochemical characterization (component composition, mass spectrometry molecular weight, post-translational modifications, epitopes, conformation, etc.), toxicity-related indicators (detection of harmful residues, virulence-related genes, virulence reversal, etc.), and efficacy-related indicators (in vitro/in vivo efficacy, quantitative humoral immunity and/or cellular immunity analysis, etc.).
Extended characterization studies for adjuvanted vaccine drug products may involve additional special considerations. The principles of extended characterization studies for aluminum-adjuvanted vaccine drug products can be found in the Technical Guidelines for Preventive Aluminum-Adjuvanted Vaccines. The studies shall include antigen, adjuvant, buffer/excipient interactions and compatibility studies, etc. The general principles for extended characterization of drug products containing other types of adjuvants are similar to those for aluminium-adjuvanted vaccines, but more characterization studies are required depending on the characteristics of the adjuvant or adjuvant system.
4. Bridging Study
Nonclinical and/or clinical bridging or confirmatory studies shall be conducted when the relationship between a specific quality attribute and safety and efficacy has not been established and differences in quality attributes between pre- and post-change products are observed. In addition, for some vaccine products, the impact of changes on safety and immunogenicity (protective effect) cannot be comprehensively reflected by pharmaceutical study alone, such as changes in some key raw materials/excipients, extension of master seed batches of attenuated live vaccines, changes in drug product dosage form, adjustment of adjuvant content, and when multiple associated changes that may affect product quality are carried out simultaneously. In such cases, nonclinical and/or clinical bridging studies shall be considered. Relevant studies may be conducted with reference to the published Technical Guidelines for Preclinical Studies of Preventive Vaccines and Technical Guidelines for Clinical Comparability Studies of Preventive Vaccines.
5. Considerations on Seasonal Influenza Vaccines
In principle, the annual updates by the vaccine manufacturers using the WHO recommended seasonal influenza vaccine virus strains are moderate changes. For routine annual updates, vaccine holders shall submit the corresponding study data of updated virus seeds, and carry out quality review and stability study review to support the annual changes of influenza strains compositions with relevant platform knowledge. The specific updated study content shall at least include: source information of virus seed lots, process of establishing the tertiary seed bank, test results of the tertiary seed batches and necessary validation data of manufacturing process; meanwhile, the holders shall submit the stability investigation plan for the current year and carry out the stability study; and provide relevant information of influenza standard substances for the current year. In addition, updated product inserts, packaging and labeling information should be provided.
The annual update of seasonal influenza vaccines is a process of dynamic adjustment based on global influenza surveillance and virological analysis and other data, and there are complex situations such as sequence replacement and type combination adjustment, etc. With the new influenza vaccines continuously on the market, there may be additional considerations for the above situations, and the specific management of changes and technical requirements are separately stipulated.
Other influenza vaccine-related changes not associated with the annual updates should be investigated in accordance with normal post-marketing changes. Due to time-sensitive influenza vaccine production, vaccine holders should make reasonable arrangements for changes unrelated to the annual updates of the vaccine strains and should not include these changes in the annual updates of the strains so as not to delay the progress of approval. At the same time, the holders shall maintain good communication with regulators regarding annual updates of the strains to ensure that influenza vaccine production occurs before the start of the influenza season.
III. Classification of Changes
According to the nature and degree of changes in the vaccine process as well as the degree and risk level of potential impact on the quality attributes, safety or immunogenicity (protective effect) of the vaccine, the changes are divided into three categories from high to low: major changes, medium changes and minor changes. Where the change may affect the safety, efficacy and quality controllability of the vaccine, it shall be approved by the drug regulatory authority under the State Council.
For some items that may result in significant differences in the quality attributes of the vaccine, which in turn affect the safety, efficacy and quality controllability of the vaccine, such as change from non-purified or whole cell (bacterial, viral, etc.) vaccines to purified or component vaccines, use of vaccines with new bacterial and viral strains, cell matrix or expression system, change to the carrier of marketed conjugate vaccines, use of new inactivators (methods) or detoxifiers (methods), and use of new adjuvants, etc. it is necessary to consider submitting new clinical trial and registration applications in accordance with the Provisions for Drug Registration and the requirements for registration classification and application dossiers of biological products.
For post-marketing changes of vaccines, the production site inspection, specification verification or sample test may be timely conducted according to relevant regulations and the need for technical review.
IV. Communication
The pharmaceutical changes of vaccines are complex and diverse. The contents of the Guidelines cannot list all the changes one by one, and the classification of changes often requires risk assessment and comprehensive judgment in combination with study results and product knowledge. The MAH is encouraged to communicate with corresponding drug regulatory authorities and technical units on key technical issues related to changes in the vaccine manufacturing process not covered by the Guidelines, such as the expected classification of changes in vaccines, the study items supporting changes and the management scheme for post-marketing changes, through communication channels in accordance with relevant requirements in the Provisions for the Administration of Post-marketing Changes of Drugs (Interim). The MAH is encouraged to communicate with corresponding regulatory authorities as soon as possible for the special circumstances of manufacturing process changes that may affect the accessibility of vaccines for immunization programs and domestic supply.
V. Categories and Technical Requirements of Pharmaceutical Changes of Vaccines
This section lists common pharmaceutical changes of vaccines, especially changes in the manufacturing process, and defines the categories of specific changes, prerequisites to be met and basic technical requirements. The classification of changes is based on the extent and risk level of the potential impact that the changes may have on the quality attributes, safety or immunogenicity (protective efficacy) of vaccines and is harmonized with relevant international guidelines. If all of the prerequisites are not met for the corresponding change, the change shall be placed in a higher category until all of the prerequisites are met (e.g., If all of the prerequisites for a medium change are not met, the change shall be a major change). Regardless of the change classification and application route, for the corresponding change items, comprehensive comparability study shall be conducted by referring to the Guidelines and included in the change control system of the quality management system.
In the Guidelines, minor changes directly involved in the items or contents in the drug registration approval document and its appendixes shall be managed as per filing (e.g., minor changes in the registration specification shall be managed as per filing).
For the convenience of application, the Common Technical Document (CTD) sections involved in each common pharmaceutical change of vaccines are indicated in the Guidelines to align with CTD application.
1. Seed batches and cell banks for production
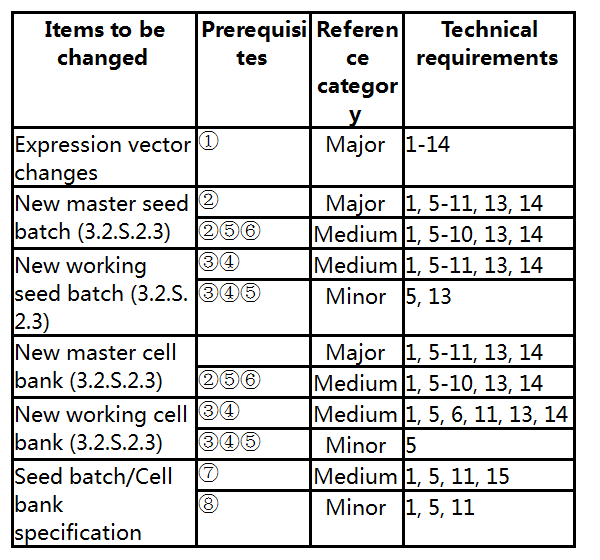
Prerequisites:
① The target gene and host cell are not changed.
② The new master seed batch/new master cell bank is prepared from a previously approved primary seed batch/cell bank or an approved master seed batch/cell bank.
③ The new working seed batch/new working cell bank is prepared from a previously approved master seed batch/master cell bank.
④ The passage of the new working seed batch/new working cell bank does not exceed the passage previously approved.
⑤ The preparation method is unchanged, and the specification of the seed batch/cell bank is tightened or unchanged.
⑥ The passage of the new master seed batch/new cell bank does not exceed the approved passage.
⑦ Where the test method is updated or changed with the edition of domestic and foreign pharmacopoeias, the specification after change meets the requirements of Chinese Pharmacopoeia.
⑧ The addition of new test items or tightening of the acceptance criteria comply with the pharmacopoeia and other relevant domestic and foreign specifications and guidelines.
Technical requirements:
1. Explain the reason for change. The content, basis and advantages of the change shall be described in detail.
2. Describe the name, source, structure and genetic characteristics of the expression vector. Describe the composition and function of the vector. Use of special vectors with limited current knowledge shall be justified and the safety and use advantages of the vectors shall be evaluated.
3. Describe the construction and screening methods of expression vectors and/or viral vectors in detail. Whether the result of enzyme digestion identification is correct or not. Provide the sequencing profile for the nucleotide sequences of the control regions at both ends of the inserted gene and the expression vector, and compare the results to indicate whether the results comply with the designed (theoretical) sequence.
4. Describe the introduction of the recombinant expression vector into host cells (bacteria) as well as the methods of monoclonal screening and confirmation in detail. Based on the risk, analyze the status of the target gene and associated control elements within the host cell (whether integrated into the chromosome or not), the copy number and the genetic stability of the host after binding to the vector. The method and expression level used to initiate and control the expression of the target gene in host cells.
5. The preparation, management and testing of seed batches and/or cell banks shall comply with the relevant requirements of “Management and quality control of bacterial and viral species for production and testing of biological products” and/or “Preparation and quality control of animal cell substrates for production and testing of biological products” in Chinese Pharmacopoeia. If applicable, the passage process, preparation method and preparation scale of seed batches/cell banks shall be described in detail. The complete certificate of analysis of seed batches/cell banks shall be provided.
6. Clarify the storage site, method and conditions of seed batches/cell banks at all levels. If involved, the study data of the passage stability of seed batches/cell banks shall be provided. The maximum allowable multiplication or passage times during scale production shall be analyzed and determined.
7. Perform process validation of three consecutive batches of commercial scale drug substance and drug product (if there is an impact on the drug product). The suitability of the seed batch/cell bank shall be confirmed by the consistency of successive batches of the product, demonstrating that the risk of contamination by extrinsic factors and variation can be avoided. For seed batches of multivalent vaccine intermediates, the MAH may appropriately reduce the number of batches studied (by bracketing, matrixing, etc.), but sufficient justification shall be provided.
8. Provide results under accelerated and/or degraded conditions for at least 3 months (or until nonconformance) of the pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product), unless otherwise required. Provide at least 3-6 months (or until nonconformance) of real-time/actual stability data for pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product). A comparability study of the stability of pre- and post-change drug substance and drug product (if there is an impact on the drug product) under accelerated and/or forced degradation and real-time/actual conditions shall be performed. Pre-change data can be historical stability testing results.
9. Develop the stability study protocol. Long-term stability studies shall be continued to confirm the hold time/shelf life of the drug substance and drug product (if there is an impact). Commitment shall be made to report nonconforming situations in long-term stability studies.
10. Nonclinical and/or clinical bridging studies shall be conducted when data from the pharmaceutical comparability study are insufficient to support a change in comparability. In case of application for exemption, there shall be sufficient reasons and basis.
11. If involved, update seed batch/cell bank specifications. Provide the basis for the change to the specification of seed batch/cell bank and the test results.
12. If involved, specify the source and characteristics of bacterial (viral) species.
13. Carry out comprehensive testing of end-of-production and/or ultra-end-of-production seed batches/cell banks, including testing on the genetic stability and microbial contamination of cells and bacterial (viral) strains during production. The testing results shall comply with the requirements of Chinese Pharmacopoeia and other relevant international guidelines.
14. Provide the pre- and post-change quality comparability study of three consecutive batches of commercial scale drug substance and drug product (if there is an impact on the drug product).
15. For methods already available in the pharmacopoeia, validation of the analytical method shall be performed prior to the initial use; a comparative study on the specification before and after the change shall be conducted; the suitability of the analytical method shall be investigated to demonstrate that the proposed analytical method is equivalent or superior to the approved method.
2. Media and raw materials for production
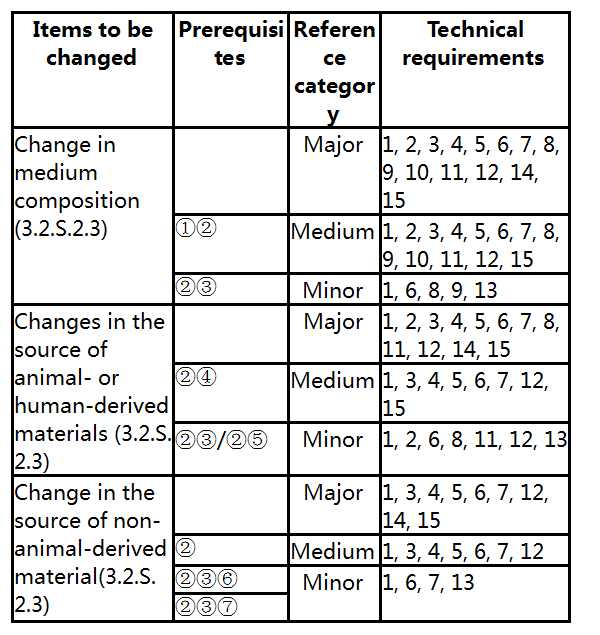
Prerequisites:
①Changes to the key components, such as addition, removal, replacement, increase, decrease and supplier change.
② No impact on the critical quality attributes of the product.
③Changes to non-key components, such as addition, removal, replacement, increase, decrease and supplier change.
④ Replacement by non-animal-derived raw materials, such as tissue or plasma-derived raw materials changed to recombinant products, replacement of animal-derived raw materials with plant-derived or synthetic raw materials, etc.
⑤Replacement by animal-derived raw materials conforming to pharmacopoeia specifications, such as newborn bovine serum.
⑥ For example, a change from salt A to salt B with a similar mechanism of action; or the type of substance is not changed, and only the supplier is changed.
⑦ Remove the antibiotics in production according to the requirements of Chinese Pharmacopoeia.
Technical requirements:
1. Explain the reason for change. The source of raw materials for production, and the change in active ingredients and the similarities and differences in the specifications before and after the change shall be specified. The quality test report shall be provided. The quality and stability of the raw materials for production shall be evaluated in combination with the test report of key raw materials. In case of changes in analytical methods, methodological validation/verification is required. If necessary, the validation may also involve the validation of virus inactivation/removal, the validation of the storage period of intermediate products, and the study on the service life of filter membrane and chromatography medium.
2. Evaluate the viral safety of animal-derived or human-derived materials, if involved. Bovine-derived substances shall have evidence of non-endemic origin and be assessed for TSE safety risk in accordance with relevant national regulations and the “Note for Guidance on Minimizing the Risk of Transmitting Animal Spongiform Encephalopathies via Human and Veterinary Medicinal Products” (EMA). Replacement of animal-derived raw materials with recombinant products is encouraged to minimize the risk of contamination with extrinsic factors.
3. Perform process validation for at least three consecutive batches of commercial scale drug substance and drug product (if there is an impact on the drug product). Pre- and post-change in-process control and product quality comparability studies shall be performed to demonstrate the comparability of pre- and post-change drug substance and drug product (if there is an impact on the drug product).
4. Revise the specification of the drug substance, and perform methodological validation for the new analytical methods, if involved.
5. Provide results under accelerated and/or forced degradation conditions for at least 3 months (or until nonconformance) of the pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product), unless otherwise required. Provide at least 3-6 months (or until nonconformance) of real-time/actual stability data for pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product). A comparability study of the stability of pre- and post-change drug substance and drug product (if there is an impact on the drug product) under accelerated and/or forced degradation and real-time/actual conditions shall be performed. Pre-change stability data can be derived from historical stability testing.
6. The raw materials for production shall meet the production requirements and comply with the provisions of “Quality Control of Raw Materials and Excipients for the Production of Biological Products” in Chinese Pharmacopoeia and relevant international guidelines.
7. In principle, the use of materials that are toxic and harmful to human body shall be avoided as far as possible in the production process. When they must be used, the removal effect of subsequent processes shall be validated. Unless the validation results indicate that the residues of process-related impurities are far below the specified requirements, or there is evidence proving the residues are within the acceptable range for humans, the test item for such residues shall normally be set at the stage of drug product testing or appropriate intermediate product control.
8. If involved, studies shall be conducted by referring to the relevant international technical guidelines, and the biological safety assessment or statement shall be provided.
9. Medium suitability test shall be carried out to analyze and verify the effect of the change in medium composition on the active ingredient.
10. The media for production shall not contain substances that may cause adverse reactions in humans, and penicillin or other β-lactam antibiotics shall not be used. If a medium containing animal-derived ingredients is replaced by a medium containing non-animal-derived ingredients or chemically defined ingredients, attention shall be paid to the effects of the medium on the cell (bacteria) growth curve, virus proliferation curve and products.
11. If involved, the trypsin used to digest cells shall be demonstrated to be free of adventitious or endogenous viral contamination. Eggs used for the preparation of embryonated eggs or embryonated egg cells shall be derived from specific pathogen free (SPF) flocks unless otherwise specified. The use of antibiotics and preservatives in the production process shall comply with the relevant requirements of Chinese Pharmacopoeia.
12. A stability study protocol shall be developed, if applicable. Long-term stability studies shall be continued to confirm the full hold time/shelf life of the drug substance and drug product (if there is an impact on the drug product). Commitment shall be made to report nonconforming situations in long-term stability studies.
13. If applicable, process verification (e.g., the batch scale covers routine production, the production process meets the predetermined in-process control standards, the product meets the specification, sterility assurance level, etc.) of at least one commercial production scale drug substance and drug product (if there is an impact on the drug product) shall be conducted, and a comparison of the in-process control and product quality before and after the change shall be conducted. The suitability of the two sources of raw materials and comparability of the drug substance and drug product (if there is an impact on the drug product) shall be demonstrated.
14. Nonclinical and/or clinical bridging studies shall be conducted when data from the pharmaceutical comparability study are insufficient to support a change in comparability. In case of application for exemption, there shall be sufficient reasons and basis.
15. Genetic stability studies of cell passages shall be performed, if applicable. The maximum allowable cell doublings or passage times during scale production shall be analyzed and determined. At the end of the production cycle, the characteristics of the host cell/vector system, such as cell viability, plasmid (target gene) copy number, extrinsic factors, restriction endonuclease map, target gene expression level and nucleic acid sequencing analysis, shall be monitored to confirm the genetic stability of cells (bacteria) during production. Comprehensive testing data of the extrinsic factors at the end of production shall be provided.
3. Preparation of Bacterial Cultures

*: For those belonging to the items already in the table, they shall be implemented according to these Guidelines; if not, communication is recommended.
Prerequisites:
① This change has no impact on the bactericidal efficacy.
② The changes to non-target components do not exceed the approved limits. No new non-target components appear.
③ No significant changes to antigens occur, and the quality of vaccines shall not be adversely affected, so that the vaccines are more suitable for commercial scale production.
④ The generation of bacteria is within the approved range.
⑤ The approved culture process parameters are not changed.
⑥ Safety indicators, such as extrinsic factors and impurity residues, are not affected.
⑦ The sensitivity of the detection of extrinsic factors is not affected.
⑧ The composition of antigens does not change obviously, and the purity of antigens is not affected.
⑨ The in-process control parameters do not affect the critical quality attributes of the product.
⑩ The change does not affect the safety and quality of the product.
⑪ The change does not affect the purification process.
⑫ The quality of the drug substance does not exceed the approved range and limits.
⑬ The change is not related to recurring deviations in production or stability concerns.
⑭ If involved, in-process control shall be tightened within the approved range. If an in-process control method and limit are replaced, the new method is a compendial method and is not a biological/immunological/immunochemical method or is not a method for testing biological active substances using biological reagents (excluding compendial microbiological test methods).
Technical requirements:
1. Explain the reasons for change, specify the specific contents of change, and describe the reasons for change and specific changes (production equipment, process route, control method of production process, acceptable range, etc.) in detail. Perform process comparison before and after change. Evaluate the rationality and feasibility of the manufacturing process.
2. If involved, specify the serum, antibiotics and other added ingredients in the culture medium. Describe whether there are changes to the raw materials (manufacturer, grade, test method and specification) and equipment for production, manufacturing process and scale, specification (analysis item, method and limit), packaging materials and containers. Pay attention to the compatibility of the performance, working principle and production capacity of the production facilities and equipment with the manufacturing process before and after the change. If involved, provide the supporting basis.
3. Conduct change process study. Provide the flow chart of the manufacturing process, indicate the process steps and process control parameters, and show the materials (material) addition link. Briefly describe the proposed manufacturing process, and specify the culture medium, bacterial fermentation/harvest process mode, scale and batch definition. If involved, conduct study to determine the fermentation process parameters (such as the inoculation ratio, temperature, pH value, stirring speed, aeration, dissolved oxygen, etc.), process control limits (such as bacterial density, viable bacterial count, induction and expression conditions, microbial contamination monitoring, antigen yield, etc.), culture cycle and harvest endpoint, harvest/pooling method, sterilization process, ultrafiltration concentration and other process parameters, detoxification process, etc. Determine indicators for discarding culture.
4. Perform process validation for at least three consecutive batches of commercial scale drug substance and drug product (if there is an impact on the drug product). The scale of the validation batch (whether it is consistent with the designed manufacturing capacity) and the analysis of the representativeness of the manufacturing process (e.g., whether it can cover the range of routine manufacturing scale) shall be clarified. Validation shall also include the analysis of continuous production batches meeting their predefined in-process controls and specifications, and analytical validation of effects of the process on the type and content of product-related impurities. If involved, validation of the detoxification effect and the storage period of intermediate products, and study on the service life of the filtration membrane and other media shall be conducted.
5. Develop the protocol for the change comparability study. Unless otherwise specified, analysis and study of the process, in-process control and quality (identification, biological activity, purity, impurities, contamination, etc.) shall be performed for at least three consecutive batches of post-change commercial scale drug substance and drug product (if there is an impact on the drug product), and a comparability study shall be performed with pre-change drug substance and drug product.
6. Provide results under accelerated and/or forced degradation conditions for at least 3 months (or until nonconformance) of the pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product), unless otherwise required. Provide at least 3-6 months (or until nonconformance) of real-time/actual stability data for pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product). A comparability study of the stability of pre- and post-change drug substance and drug product (if there is an impact on the drug product) under accelerated and/or forced degradation and real-time/actual conditions shall be performed. Pre-change stability data can be derived from historical stability testing.
7. Develop the stability study protocol. Make a commitment to continue long-term stability studies to confirm the full hold time/shelf life of the drug substance and drug product (if there is an impact on the drug product). Commitment shall be made to report nonconforming situations in long-term stability studies.
8. Nonclinical and/or clinical bridging studies shall be conducted when data from the pharmaceutical comparability study are insufficient to support a change in comparability. In case of application for exemption, there shall be sufficient reasons and basis.
9. If the change results in a change to the production generation of bacteria, the study on extrinsic factors and genetic stability at the end of production shall be conducted according to the requirements of Chinese Pharmacopoeia and other relevant international guidelines. Bacteria shall be tested for purity and total viable bacteria count at the end of production.
10. If involved, the raw materials for production shall comply with the provisions of Chinese Pharmacopoeia and relevant international guidelines. The use of human- or animal-derived raw materials in the production process shall be avoided as far as possible. If they must be used, the viral safety of animal- or human-derived materials shall be evaluated. Bovine-derived substances shall have evidence of non-endemic origin and be assessed for BSE/TSE safety risk in accordance with relevant national regulations and the “Note for Guidance on Minimizing the Risk of Transmitting Animal Spongiform Encephalopathies via Human and Veterinary Medicinal Products” (EMA). Replacement of animal-derived raw materials with recombinant products is encouraged to minimize product safety risks.
11. For bacterial polysaccharides and component vaccines, the consistency of the target product shall be validated by the bacterial growth state and the level of polysaccharide/protein in the final production, including by identification and structure elucidation.
12. Clarify the rationale for classifying the changes as medium or minor.
13. The use of organic solvents in the production process and the residue limits shall be strictly in accordance with the provisions of “Determination of Residual Solvents” in Chinese Pharmacopoeia and ICH, the use of Class I solvents shall be avoided, and the use of Class II solvents shall be limited. If organic solvents or other substances are used for extraction, purification or inactivation treatment, the subsequent purification process of the product shall ensure that the organic solvents or other substances in the product can be effectively removed.
14. For bacterial vaccines that need grinding treatment after harvest, such as BCG vaccines, if the grinding method changes, the effect on the number of viable bacteria and the activity of bacteria shall be paid attention to.
15. For toxin vaccines, in addition to focusing on the detoxification effect of the detoxification process, attention shall also be paid to the influence on the antigen yield and immunogenicity to ensure the comparability of intermediate products.
16. Detail the changes to the manufacturing process control parameters and ranges. If applicable, the comparability of pre- and post-change manufacturing process control parameters and ranges shall be demonstrated.
17. If applicable, process verification (e.g., the batch scale covers routine production, the production process meets the predetermined in-process control standards, the product meets the specification, etc.) of at least one commercial production scale drug substance and drug product (if there is an impact on the drug product) shall be conducted. A comparative analysis of the in-process control and batch release data shall be performed.
18. If involved, single-use systems shall have supplier quality assurance/quality system and core validation documentation (including sterilization validation). The study on the single-use system shall be conducted in combination with product production, including chemical compatibility, adsorption capacity, bacterial challenge, particulate matter, extractables and/or leachables, integrity, etc. If the type and content of extractables and/or leachables change from that before the change, the effect of such change on the manufacturing process (including downstream process) and the product itself shall be assessed, and whether the newly emerged extractables and/or leachables will interact with intermediate products or drug substance shall be investigated.
19. If involved, risk assessment of contamination by foreign bacteria and extrinsic factors shall be conducted.
20. If necessary, special safety test (e.g., local irritation test) shall be conducted for injections.
21. Provide information that the in-process control does not affect the critical quality attributes.
22. If involved, provide the updated specification of the drug substance, including the test items and analytical methods. If applicable, provide the updated analytical methods and methodological validation data.
23. If involved, specify the information of the equipment to be changed, and compare the similarities and differences of the operating principles and key technical parameters of the equipment before and after the change. If involved, the manufacturing process and/or equipment after change shall be fully validated, including the validation of aseptic production and sterilization process.
4. Preparation of Virus Cultures
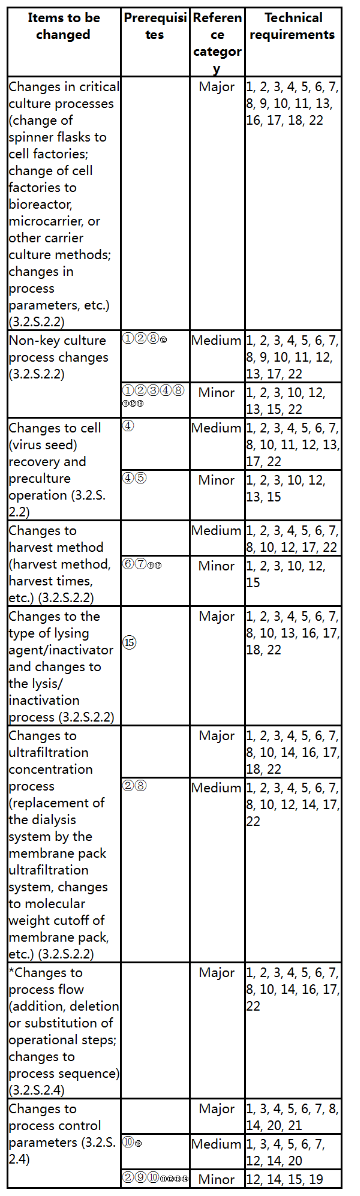
*: For those belonging to the items already in the table, they shall be implemented according to these Guidelines; if not, communication is recommended.
Prerequisites:
① No impact on viral removal and/or inactivation of the process.
② Non-target components do not exceed the approved limits. No new non-target components appear.
③ No significant changes to antigens occur, and the quality of the product shall not be adversely affected, so that the vaccines are more suitable for commercial scale production.
④ The generation of the virus is within the approved range.
⑤ The approved culture process parameters are not changed.
⑥ Safety indicators, such as extrinsic factors and impurity residues, are not affected.
⑦ The sensitivity of the detection of extrinsic factors is not affected.
⑧ The composition of antigens does not change obviously, and the purity of antigens is not affected.
⑨ The in-process control parameters do not affect the critical quality attributes of the product.
⑩ The change does not affect the safety and quality of the product.
⑪The change does not affect the purification process.
⑫ The quality of the drug substance does not exceed the approved range and limits.
⑬ The change is not related to recurring deviations in production or stability concerns.
⑭ If involved, the process control range shall be tightened within the approved range. If an in-process control method and limit are replaced, the new method is a compendial method and is not a biological/immunological/immunochemical method or is not a method for testing biological active substances using biological reagents (excluding compendial microbiological test methods).
⑮ The lysing agent/inactivator is what is used in marketed similar products.
Technical requirements:
1. Explain the reason for change and specify the specific contents of change. Perform process comparison before and after change. Evaluate the rationality and feasibility of the manufacturing process.
2. If involved, specify the serum, antibiotics and other added ingredients in the culture medium. Describe whether there are changes to the raw materials (manufacturer, grade, test method and specification) and equipment for production, manufacturing process and scale, specification (analysis item, method and limit), packaging materials and containers. If involved, provide the supporting basis.
3. Conduct change process study. Provide a flow chart of the manufacturing process (from the initial inoculum to the final harvest operation), including all steps (i.e., unit operations) and intermediate products, indicate the process steps and in-process control parameters, and show the materials (material) addition. Briefly describe the proposed manufacturing process, and specify the definitions of culture medium, cell culture mode, scale and batch. If involved, study to determine the main process parameters and control range of viral species expansion (such as the MOI, temperature, pH, stirring speed, aeration, dissolved oxygen, etc. of virus inoculation), in-process control requirements (such as cell density, cell viability, virus titer, virus proliferation curve, etc.), culture cycle, harvest method, inactivation process, etc. Determine indicators for discarding culture. If involved, the control cell culture container, culture conditions and test items shall be specified.
4. Process validation shall be conducted for at least three consecutive batches of commercial production scale drug substance and drug product (if there is an impact on the drug product). The scale of the validation batch (whether it is consistent with the designed manufacturing capacity) and the analysis of the representativeness of the manufacturing process (e.g., whether it can cover the range of routine manufacturing scale) shall be clarified. Validation shall also include the analysis of consecutive production batches meeting their predefined in-process controls and specifications, analytical validation of the effect of the process on the type and content of related impurities, and validation of the storage period of intermediate products. If involved, the virus inactivation/removal effect, lytic effect, and Study on the service life of filtration membrane the study on the service life of filter membrane and other media may also be validated.
5. Develop the protocol for the change comparability study. Unless otherwise specified, manufacturing process, in-process controls and quality analyses and studies (identification, biological activity, purity, impurities, contamination, etc.) will be performed on at least three consecutive batches of post-change commercial scale drug substance and drug product (if there is an impact on the drug product), and pre- and post-change comparability studies will be performed. For attenuated live vaccines, it is necessary to analyze whether the changes will lead to changes in virulence, etc.
6. Provide results under accelerated and/or forced degradation conditions for at least 3 months (or until nonconformance) of the pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product), unless otherwise required. Provide at least 3-6 months (or until nonconformance) of real-time/actual stability data for pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product). A comparability study of the stability of pre- and post-change drug substance and drug product (if there is an impact on the drug product) under accelerated and/or forced degradation and real-time/actual conditions shall be performed. Pre-change stability data can be derived from historical stability testing.
7. Develop the stability study protocol. Make a commitment to continue long-term stability studies to confirm the full hold time/shelf life of the drug substance and drug product (if there is an impact on the drug product). Commitment shall be made to report nonconforming situations in long-term stability studies.
8. Nonclinical and/or clinical bridging studies should be conducted when data from the pharmaceutical comparability study are insufficient to support comparability. In case of application for exemption, there shall be sufficient reasons and basis.
9. If the change results in the change of cell substrate and/or virus production generation, adventitious agents and genetic stability study at the end of production should be conducted according to the requirements of Chinese Pharmacopoeia and other applicable international guidelines.
10. If involved, the raw materials for production shall comply with the provisions of Chinese Pharmacopoeia and relevant international guidelines. The use of human- or animal-derived raw materials in the production process shall be avoided as far as possible. If they must be used, the viral safety of animal- or human-derived materials shall be evaluated. Bovine-derived substances shall have evidence of non-endemic origin and be assessed for BSE/TSE safety risk in accordance with relevant national regulations and the “Note for Guidance on Minimizing the Risk of Transmitting Animal Spongiform Encephalopathy Agents via Veterinary Medicinal Products” (EMA). Replacement of animal-derived raw materials with recombinant products is encouraged to minimize product safety risks.
11. Changes in culture methods may cause changes in the genetic sequence and/or virulence (e.g., neurovirulence) of certain viruses. The effect of the changes on virulence should be fully evaluated and appropriate studies should be conducted.
12. Clarify the rationale for classifying the changes as medium or minor. Describe the reason for the change and specific changes in detail (e.g., production equipment, process route, control method of production process, acceptable range, etc.).
13. The use of organic solvents in the production process and the residue limits shall be strictly in accordance with the provisions of “Determination of Residual Solvents” in Chinese Pharmacopoeia and ICH, the use of Class I solvents shall be avoided, and the use of Class II solvents shall be limited. If organic solvents or other substances are used for extraction, purification or inactivation treatment, the subsequent purification process of the product shall ensure that the organic solvents or other substances in the product can be effectively removed.
14. Detail the changes to the manufacturing process control parameters and ranges. If applicable, the comparability of pre- and post-change manufacturing process control parameters and ranges shall be demonstrated.
15. If applicable, process verification of at least one commercial production scale drug substance and drug product (if there is an impact on the drug product) shall be conducted, including the batch scale covers routine production, the production process meets the predetermined in-process control standards, the product meets the specification, the comparative analysis of process control and batch release data should be conducted.
16. If involved, the single-use system should have supplier quality assurance/quality system and core validation documentation (including sterilization validation). The study on the single-use system shall be conducted in combination with product production, including chemical compatibility, adsorption capacity, bacterial challenge, particulate matter, extractables and/or leachables, integrity, etc. If the type and content of extractables and/or leachables change from that before the change, the effect of such change on the manufacturing process (including downstream process, such as viral inactivation) and the product itself should be further assessed, and whether the newly emerged extractables will interact with intermediate products or drug substance should be investigated.
17. If involved, testing and risk assessment of adventitious agent contamination should be conducted.
18. If necessary, special safety test (e.g., local irritation test) shall be conducted for injection.
19. Provide information that the in-process control does not affect the critical quality attributes.
20. If involved, provide the updated specification of the drug substance, including the test items and analytical methods. If applicable, provide the updated analytical methods and methodological validation data.
21. When virus inactivation/removal treatment is involved in the production process, the specific steps and parameters of the inactivation/removal process shall be determined and process validation shall be performed to ensure the inactivation/removal effect.
22. If involved, specify the information of the equipment to be changed, and compare the similarities and differences of the operating principles and key technical parameters of the equipment before and after the change. If involved, the manufacturing process and/or equipment after change shall be fully validated, including the validation of aseptic production and sterilization process.
5. Preparation of Genetically Engineered Cell Cultures
The risks of cell culture preparation process changes in genetically engineered recombinant protein vaccines are similar to those of other recombinant therapeutic biological products. Therefore, the classification of changes and technical requirements may refer to the relevant parts of Technical Guidelines for Pharmaceutical Change Studies of Marketed Biological Products (Interim).
6. Antigen Purification, Modification and Processing
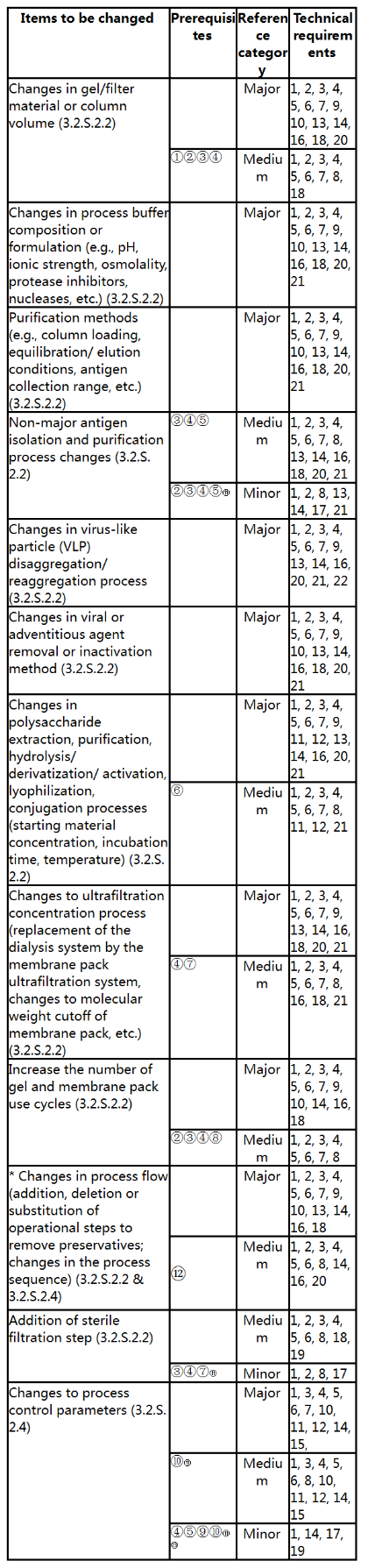
*: For those belonging to the items already in the table, they shall be implemented according to these Guidelines; if not, communication is recommended.
Prerequisites:
① Upgrade or replacement of the same type filler.
② No significant changes to antigens occur, and the quality of the product shall not be adversely affected, so that the vaccines are more suitable for commercial scale production.
③ No impact on viral removal and/or inactivation.
④ Non-target components do not exceed the approved limits. No new non-target components appear.
⑤ The possibility of influence on the quality of antigen is very small, and the quality of antigen does not exceed the approved limit.
⑥ The characteristics of polysaccharide-protein conjugates are not affected.
⑦ No effect on antigen purity.
⑧ Increase the number of gel or membrane pack use cycle according to the approved protocol.
⑨ If tightening of the process control range is involved, it should be tightened within the approved range. If the replacement of in-process control method and limit are involved, the new method is a compendial method and is not a biological/immunological/immunochemical method or is not a method for testing biological active substances using biological reagents (excluding compendial microbiological test methods).
⑩ The change does not affect the safety and quality of the product.
⑪ The change is not related to recurring deviations in production or stability concerns.
⑫ The preservatives are removed according to the requirements of Chinese Pharmacopoeia, and the antigen is not affected.
⑬ In-process parameters do not impact critical quality attributes (e.g. content, impurities, any critical physicochemical characteristics, etc.).
Technical requirements:
1. Describe the reasons for the change and the specific changes in detail (including production equipment, process route, in-process control parameters and limits, etc.). Provide the flow chart of manufacturing process, indicate the process steps and process control parameters, show the materials (material) addition link, and briefly describe the proposed manufacturing process.
2. Make clear the raw materials (manufacturer, grade, test method and specification) and equipment for production, production process and scale, specification (analysis item, method and limit), packaging materials and containers, and provide supporting data if there is any change. If the main raw materials for production are self-prepared by recombinant technology or biological/chemical synthesis technology (such as enzyme, affinity antibody, chemical conjugate, etc.), detailed production process and quality study data shall be provided.
3. Perform process validation of at least three consecutive batches of commercial scale drug substance and drug product (if there is an impact on the drug product). The scale of the validation batch (whether it is consistent with the designed manufacturing capacity) and the analysis of the representativeness of the manufacturing process (e.g., whether it can cover the range of routine manufacturing scale) shall be clarified. Validation should also include the analysis of continuous production batches meeting their predefined in-process controls and specifications, and analytical validation of effects of the process on the type and content of related impurities. If involved, the validation of the number of cycles of chromatographic fillers, the virus/bacteria inactivation/removal effect, the storage period of intermediate products, and the study on the service life of filter membrane and other media may also be validated. Where applicable, procedures for the transfer of intermediate products in each step, equipment, area and building, as well as transport and storage condition should be provided.
4. Develop the protocol for the change comparability study. Unless otherwise specified, analysis and study of the process, in-process control and quality (identification, biological activity, purity, impurities, contamination, etc.) shall be performed for at least three consecutive batches of post-change commercial scale drug substance and drug product (if there is an impact on the drug product), and a comparability study shall be performed with pre-change drug substance and drug product. For vaccines whose structure can be well characterized (e.g., genetically engineered vaccines), the impact of the change on post-translational modifications of the recombinant protein and/or on the higher order structure of the protein should be analyzed; For live attenuated vaccines, it is necessary to analyze whether the changes will lead to changes in virulence, etc. In order to ensure the comparability of the changes, if the salting-out method of toxin vaccines is changed, the precipitate amount, hemagglutination (copurified pertussis), proportion of pertussis components (copurified pertussis) and total protein content shall be mainly investigated, and attention shall be paid to the effect of the changes on antigen yield and immunogenicity to ensure the comparability of intermediate products. For some toxin vaccines and genetic engineered recombinant protein vaccines, if the sonication process is changed, attention should be paid to the effect on the solubility of antigen. If necessary, the characterization study on the antigen structure after process change should be conducted to ensure the consistency of the antigen structure before and after process change. For VLP vaccines, if the depolymerization/repolymerization process is changed, in addition to conducting the study on conventional recombinant protein products, the depolymerization/repolymerization effect before and after the change should also be compared.
5. Provide results under accelerated and/or forced degradation conditions for at least 3 months (or until nonconformance) of the pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product), unless otherwise required. Provide at least 3-6 months (or until nonconformance) of real-time/actual stability data for pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product). A comparability study of the stability of pre- and post-change drug substance and drug product (if there is an impact on the drug product) under accelerated and/or forced degradation and real-time/actual conditions shall be performed. Pre-change stability data can be derived from historical stability testing.
6. Develop the stability study protocol. Make a commitment to continue long-term stability studies to confirm the full hold time/shelf life of the drug substance and drug product (if there is an impact on the drug product). Commitment shall be made to report nonconforming situations in long-term stability studies.
7. Nonclinical and/or clinical bridging studies should be conducted when data from the pharmaceutical comparability study are insufficient to support comparability. In case of application for exemption, there shall be sufficient reasons and basis.
8. Analyze the impact of the change on the quality of the drug substance (or the impact on the quality of the drug product) based on the risk assessment, and justify the classification of the change as medium or minor.
9. If necessary, special safety test (e.g., local irritation test) shall be conducted for injection.
10. When virus inactivation/removal treatment is involved in the production process, the specific steps and parameters of the inactivation/removal process shall be determined to ensure the inactivation/removal effect.
11. For chemical coupling modified antigen, the basis for determining the process parameters to be changed shall be provided, such as the addition amount and concentration of each component of hydrolysis/derivation/activation/binding reaction system (sugar concentration, addition amount of various components such as activator/condensing agent, etc.), reaction conditions, etc. Provide the main process performance indicators of hydrolysis/derivation/activation/conjugation steps, such as: derivation rate, modification degree, overall yield, free polysaccharide, free protein, polysaccharide-protein ratio, molecular size, etc. Provide additional quality attribute comparison data, such as unbound modifiers, interpretation of NMR spectra of polysaccharides and polysaccharide derivatives (proportion of C-polysaccharides, polysaccharide integrity). If necessary, carry out the study on the kinetic curve of hydrolysis/derivation/activation/binding. Comparative studies on the immunogenicity in animals should be conducted.
12. If the extraction method of polysaccharide vaccine is changed, the polysaccharide structure should be compared when necessary in addition to the conventional comparability study.
13. If involved, the single-use system should have supplier quality assurance/quality system and core validation documentation (including sterilization validation). The disposable system shall be validated in combination with production, including chemical compatibility, adsorption capacity, bacterial challenge, particulate matter, extractables and/or leachables (long-term storage), integrity, etc. If the type and content of extractables and/or leachables change from that before the change, the effect of such change on the manufacturing process (including downstream process, such as viral inactivation) and the product itself should be further assessed, and whether the newly emerged extractables will interact with antigen should be investigated.
14. Detail the changes to the manufacturing process control parameters and ranges. If applicable, the comparability of pre- and post-change manufacturing process control parameters and ranges shall be demonstrated.
15. If applicable, provide updated drug substance specifications, including test items, analytical procedures, and acceptance criteria, and updated method suitability or validation data.
16. If involved, specify the name, main operating parameters and control range of each process step including separation and purification of active ingredients. Describe all steps and intermediate products and relevant information for each stage (e.g., volume, pH, critical processing step times, hold times, temperature and elution profiles, storage of intermediate products, etc.). Specify the type of chromatography purification process medium, main parameters related to chromatographic column (main process parameters of sample injection, equilibration, elution and other steps) and peak harvesting conditions/collection range; pore size of filter or ultrafiltration membrane, buffer composition; total protein concentration, inactivator/lyser concentration and inactivation/lysis time in the inactivation or lysis process; main process parameters of virus-like particle depolymerization and repolymerization; main process parameters of purification, hydrolysis/derivation/activation/conjugation of polysaccharides and protein carriers of polysaccharide vaccine and polysaccharide-protein conjugate vaccine, etc.
17. If applicable, process verification of at least one commercial scale drug substance and drug product (if there is an impact on the drug product) should be performed, including the following: the batch scale covers routine production, the production process meets predetermined in-process control standards, and the product meets the specification, with comparison of in-process control and batch analysis data.
18. If involved, risk assessment data of adventitious agent contamination should be provided.
19. Provide information that the in-process control does not affect the critical quality attributes.
20. If involved, adequate validation of the manufacturing process and/or equipment after change, including validation of aseptic manufacturing and sterilization processes should be performed.
21. If involved, provide information and evidence that the raw materials for production are absence of potential risk of BSE/TSE, such as the name of the supplier, the species and tissue from which the raw materials are derived, the country of origin of the animals, and the use of the raw materials. The use of organic solvents in the production process and the residue limits shall be strictly in accordance with the provisions of “Determination of Residual Solvents” in Chinese Pharmacopoeia and ICH, avoiding the use of Class I solvents and limiting the use of Class II solvents. If organic solvents or other substances are used for extraction, purification or inactivation treatment, the subsequent purification process shall ensure that the organic solvents or other substances in the product can be effectively removed, and the removal process shall be validated.
22. For the depolymerization/repolymerization of VLP vaccines, the justification for the proposed change of process parameters should be provided, such as: concentration of dissociating agent, composition of buffer system, incubation and diafiltration time, etc. Main process control of depolymerization/repolymerization step, such as depolymerization effect, reassembly effect, protein concentration, purity, percentage of intact monomer, overall yield, impurity removal, should be provided. Comparative data for additional quality attributes such as particle morphology, protein oligomer content, particle size and distribution, disulfide bonds, deamidation should be provided. Comparative studies on the immunogenicity of vaccine finished product in animals should be conducted. Comparability studies on the stability of drug substance and finished product should be carried out.
7. Other Changes (Drug Substance)

Prerequisites:
① The new manufacturing site/plant/production line is the approved drug substance manufacturing site of similar products, and the site has manufacturing experience of similar products.
② Production line replication (excluding substantial changes in manufacturing process and/or process control).
③ The new manufacturing site/plant/production line is under the same quality assurance/quality control (QA/QC) system as the current manufacturing site.
④ The change of non-target components does not exceed the approved limits. No new non-target components appear.
⑤ The change does not affect the purification process.
⑥ The quality of the drug substance does not exceed the approved range and limit of the specification.
⑦ Only the fermentation scale is changed, but the same bioreactor is still used.
⑧ The change of equipment will not affect other equipment. There is no change in the material in contact with the drug substance or operating principle of the equipment before and after the change.
⑨ Process parameters should not exceed the validated range after equipment change. The sterility level/microbial limit is not reduced.
⑩ Equivalent equipment (with similar equipment design, same operating principle, same or higher grade product contact materials. Equivalent equipment shall provide product of the same quality as the product processed by the previous equipment) is used to replace the current equipment.
⑪ Disposable reservoir bags, packaging materials and containers, filter membranes and so forth before and after the change are equivalent (including transport stability study, etc.) The change does not increase the risk of extractables/leachables. Packaging materials, containers, and the sterilization process of the filter membrane are unchanged.
⑫ There is no change in packaging materials and storage conditions for intermediate products or drug substance.
⑬ Stability study is performed as per the approved stability study protocol.
⑭ Complete long-term stability data covering the proposed storage period are available from at least three consecutive batches of commercial scale intermediate products and drug substance, and no significant changes have been observed in stability study.
⑮ The change is not related to recurring deviations in production or stability concerns.
⑯ The amount of raw materials varies linearly with the scale; Process parameters remain within the approved range or vary linearly with scale.
⑰ The change does not affect the effectiveness of the viral clearance and/or inactivation process.
⑱ Tightening within the proposed temperature range.
Technical requirements:
1. Describe the reason for the change, and describe the content of change in detail (production equipment, process route, control method of production process, acceptable range, etc.).
2. Make clear the raw materials (manufacturer, grade, test method and specification) and equipment for production, production process and scale, specification (analysis item, method and limit), packaging materials and containers. In case of any change, provide supporting data.
3. Perform process validation of at least three consecutive batches of commercial scale drug substance and drug product where applicable (if there is an impact on the drug product). The scale of the validation batch (whether it is consistent with the designed manufacturing capacity) and the analysis of the representativeness of the manufacturing process (e.g., whether it can cover the range of routine manufacturing scale) shall be clarified. Validation should also include the analysis of continuous production batches meeting their predefined in-process controls and specifications, and analytical validation of effects of the process on the type and content of related impurities. If involved, the virus/bacteria inactivation/removal effect, the storage period of intermediate products, and the study on the service life of filter membrane and other media may also be validated. Where applicable, procedures for the transfer of intermediate products in each step, equipment, area and building and storage condition should be provided.
4. Develop the protocol for the change comparability study. Unless otherwise specified, process, in-process controls and quality analyses will be performed on at least three consecutive batches of post-change commercial scale drug substance and drug product (if there is an impact on the drug product), and pre- and post-change comparability studies will be performed.
5. Provide results under accelerated and/or forced degradation conditions for at least 3 months (or until nonconformance) of the pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product), unless otherwise required. Provide at least 3-6 months (or until nonconformance) of real-time/actual stability data for pre- and post-change commercial scale drug substance and drug product (if there is an impact on the drug product). A comparability study of the stability of pre- and post-change drug substance and drug product (if there is an impact on the drug product) under accelerated and/or forced degradation and real-time/actual conditions shall be performed. Pre-change stability data can be derived from historical stability testing. If involved, transport stability studies should be performed.
6. Develop the stability study protocol. Long-term stability studies of the drug substance and drug product (if there is an impact on the drug product) shall be continued to confirm the hold time/shelf life of the drug substance and drug product. Commitment shall be made to report nonconforming situations in long-term stability studies.
7. Nonclinical and/or clinical bridging studies should be conducted when data from the pharmaceutical comparability study are insufficient to support comparability. In case of application for exemption, there shall be sufficient reasons and basis.
8. Explain the rationale for classifying the changes as major, medium or minor.
9. If involved, conduct change process study. Provide the flow chart of the manufacturing process, indicate the process steps and process control parameters, and show the materials (material) addition link. Briefly describe the proposed manufacturing process.
10. If the change results in the change of bacterial (viral) species/cell generation, adventitious agents and genetic stability study at the end of production should be conducted according to the requirements of Chinese Pharmacopoeia and other applicable international guidelines. Bacteria shall be tested for purity and total viable bacteria count at the end of production.
11. If involved, update the information of the equipment, and provide the comparison information of the similarities and differences of the operating principles and key technical parameters of the new equipment and the replaced equipment. Perform validation or revalidation on the equipment. If involved, provide the leaching/extraction study data of equipment.
12. If involved, provide process flow description, revise the information of control strategy of critical process steps and intermediate products, and compare the process and in-process controls before and after the change.
13. Perform manufacturing process verification of at least one batch of drug substance at commercial production scale (e.g., the batch scale covers routine production, the production process meets the predetermined in-process control standards, the product meets the specification, etc.), and compare the in-process control and batch analysis data before and after the change.
14. If necessary, special safety test (e.g., local irritation test) shall be conducted for injection.
15. If involved, the feasibility of shared equipment in contact with the product should be verified and specific procedures for cross-switching should be described.
16. If involved, the single-use system should have supplier quality assurance/quality system and core validation documentation. The MAH shall validate the disposable system in combination with vaccine production, including chemical compatibility, adsorption capacity, bacterial challenge, particulate matter, extractables and/or leachables, integrity, etc. If the type and content of extractables and/or leachables change from that before the change, the effect of such change on the manufacturing process (including downstream process, such as viral inactivation) and the product itself should be further assessed, and whether the newly emerged extractables and/or leachables will interact with intermediate products or drug substance should be investigated.
17. Describe the updated packaging materials and containers in detail (such as appearance, composition, raw materials in direct contact with drug packaging materials and containers, etc.), and conduct the compatibility study of packaging materials. Demonstrate that the updated packaging materials and containers are at least similar to the approved packaging materials and containers with respect to the respective characteristics.
18. Update the specifications (including analytical methods) for drug packaging materials and containers, explain the basis of the specifications, and compare the specifications or test methods before and after the change.
19. Compatibility studies should be conducted for the change of other contact materials used in the production, such as containers of intermediate products used in the production.
20. A justification for the change in storage conditions and/or shelf life should be provided. Unless otherwise specified, intermediate products should proceed to subsequent processing steps in a continuous production process. Long-term stability studies of the drug substance and drug product (if there is an impact on the drug product) shall be continued to confirm the hold time/shelf life of the drug substance and drug product if applicable. Commitment shall be made to report nonconforming situations in long-term stability studies.
21. Full real-time/real-world stability studies are generally completed for at least three consecutive batches of commercial scale intermediate products or drug substance. Each intermediate product is stored under the most demanding storage conditions to be set (including the most demanding temperature, the longest storage period, the most likely risk of potential contamination, etc.), and at least three batches of the finished vaccines made from these intermediate products should be subjected to accelerated and/or stress stability and real-time/real-world stability validation. Data should be specified on whether the change adversely affects the quality of intermediate products or drug substance. If relaxation of the storage conditions or extension of the storage period of intermediate products is involved, the supporting materials showing that there is no adverse effect on the antigen should be provided. If involved, appropriate transport stability validation studies, including stability studies at extreme temperatures, etc., should be conducted.
22. If applicable, make appropriate revision on the contents such as specifications and production and inspection procedures.
23. If there is change in stability study protocol, provide an updated post-approval stability study protocol and commitment, along with a justification for the change.
24. In principle, intermediate products and drug substance should proceed to subsequent processing steps in a continuous production process. When temporary storage is required due to the interval before obtaining test results, appropriate storage methods and conditions should be selected, degradation products and aggregates that may affect the efficacy and safety should be tested, and acceptance criteria should be established.
25. The storage period and/or storage conditions of intermediate products and drug substance are determined based on the results of long-term stability testing after the change, and extrapolation of the results for the vaccine bulk is not applicable.
26. Specify the information of samples for stability study, and describe the batch number, production date and storage container of samples. Summarize completed stability studies.
27. The comparability study of stability under the two storage conditions before and after the change should be conducted, and the study items should be comprehensive.
28. Commitment shall be made to use the drug substance at the end of storage period to be changed to prepare the drug product, and complete the long-term stability study data covering the full shelf life of the drug product.
29. If the analytical method is changed, the new analytical method should be detailed and validated. Demonstrate that the new analytical method is comparable or superior to the approved analytical method.
30. If involved, provide information and evidence that the raw materials for production are absence of potential risk of BSE/TSE, such as the name of the supplier, the species and tissue from which the raw materials are derived, the country of origin of the animals, and the use of the raw materials. The use of organic solvents in the production process and the residue limits shall be strictly in accordance with the provisions of “Determination of Residual Solvents” in Chinese Pharmacopoeia and ICH, avoiding the use of Class I solvents and limiting the use of Class II solvents. If organic solvents or other substances are used for extraction, purification or inactivation treatment, the subsequent purification process shall ensure that the organic solvents or other substances in the product can be effectively removed, and the removal process shall be validated.
31. If involved, adequate validation of the manufacturing process and/or equipment after change, including validation of aseptic manufacturing and sterilization processes should be performed.
32. If necessary, animal safety and efficacy evaluation shall be conducted according to the characteristics of the product.
8. Manufacturing Process of Finished Products

*: For those belonging to the items already in the table, they shall be implemented according to these Guidelines; if not, communication is recommended.
Prerequisites:
① There is no major change in the manufacturing process.
② The approved indications, usage and dosage and target population remain unchanged.
③ Reduce or increase the additional deliverable volume.
④ For example, the sterile filtration step in the preparation of semi-finished products is added, or the in-line sterile filtration step is added in the filling process.
⑤ The change does not affect the preparation point of each component of the semi-finished product, and has no significant effect on the antigen content and overall quality of the vaccine.
⑥ Within the approved validation process range, the change has no impact on the antigen content and overall quality of the vaccine.
⑦ Within the validated lyophilization parameters, the lyophilization curve is basically unchanged.
⑧ The change is not related to recurring deviations in production or stability concerns.
⑨ The test method is the same or only slightly changed.
⑩ The preservatives are removed according to the requirements of Chinese Pharmacopoeia, and the antigen is not affected.
⑪ The texture and types of packaging materials and containers remain unchanged.
⑫ The adjuvant type is aluminum-containing adjuvant.
⑬ The quality characteristics of the aluminium adjuvant do not change as a result of the change.
⑭ There is no change to the manufacturer and the process parameters do not exceed the validated ranges after the change.
⑮ If involved, the in-process control range shall be tightened within the approved range. If an in-process control method and limit are replaced, the new method is a compendial method and does not involve a biological/immunological/immunochemical method or is not a new method for testing biological active substances using biological reagents (excluding compendial microbiological test methods).
⑯ No safety and quality issues are involved.
⑰ Critical quality attributes are not affected by in-process control parameters.
⑱ Sterility levels/microbial limit levels of the drug product are not affected.
⑲ The changes do not involve the sterility level of the diluent.
⑳ It refers to water for injection or buffered saline solutions, i.e., which do not contain the active ingredient (e.g., adjuvants, etc.) ; there is no change in the composition of the diluent.
There is no significant impact on the quality of the reconstituted drug product.
Technical requirements:
1. Elaborate the specific contents of the change in detail. Provide the necessity, scientificity and rationality of the change.
2. Detail the composition information of new batch formula. Provide the basis for determining the new formulation, including literature information, study information (including the study contents of formulation design, formulation screening and optimization, formulation determination, etc.), and comment on whether the adjuvant, stabilizer, buffer and excipient in the formulation affect the immunogenicity and safety of the vaccine. The batch formula should include a list of all ingredients used in the manufacturing process of the finished product, the amount of each ingredient in each batch, including overages, and the sources and specifications of each ingredient.
3. Provide the process flow chart to show the steps of sample entering the process. Provide the preparation method of semi-finished product, critical process parameters and in-process control range. The formulation process description shall reflect the concept of “point formulation”. The batch size should be specified. Carry out the scale production process study, and elaborate on the new process and packaging operation that directly affect the quality of vaccine.
4. Carry out commercial scale manufacturing process validation for at least three consecutive batches of the new manufacturing process. It should be clarified whether the scale of the validation batch is consistent with the designed manufacturing capacity and an analysis that is representative of the manufacturing process should be performed (e.g., whether the range of routine manufacturing scale can be covered; whether worst-case process conditions can be represented). Validation should include the analysis of consecutive production batches meeting their predefined in-process control standards and specifications; Aseptic process validation; Validation of solution homogeneity; Lyophilization process validation (if applicable) ; Adsorption kinetics curve (if applicable) ; et al.
5. Provide the study protocol of change comparability, and comprehensively carry out the comparability study on the manufacturing process and quality of vaccine diluent (if involved), adjuvant (if involved) and drug product at commercial production scale before and after the change based on the risk.
6. If involved, conduct standard study and necessary standard revision in combination with the impact of the change on the quality of the vaccine. Detail the analytical methods and carry out the necessary methodological validation.
7. Detail the information of packaging materials and containers. If involved, studies on the integrity of the packaging materials and container closure should be performed. If the type of immediate packaging materials and containers is changed, the stability study of preparation and compatibility study of packaging materials should also be conducted.
8. Provide results under accelerated and/or forced degradation conditions for at least 3 months (or until nonconformance) of the pre- and post-change commercial scale drug product, unless otherwise required. Provide at least 3-6 months (or until nonconformance) of real-time/actual stability data for pre- and post-change commercial scale drug product. Comparability studies are performed for pre- and post-change drug product stability under accelerated and/or forced degradation and real-time/actual conditions. Pre-change data can be historical stability testing results. For multi-dose vaccines, a commitment should be made to provide in-use stability data at the end of the shelf life of the post-change drug product to demonstrate consistent quality of the post-change product over the actual multiple use periods. If involved, transport stability studies should be performed. A change in the strength (concentration, volume) of the drug product may render it impossible to support full shelf life approval with limited post-change stability study data. Stability study data of the post-change drug product covering the proposed shelf life should be provided to support full shelf life approval.
9. Commitment shall be made to conduct long-term stability studies to confirm the full shelf life of the vaccine under normal storage conditions. Commitment shall be made to report any rejections in the long-term stability study.
10. If involved, special safety tests (e.g., local irritation test, etc.) shall be conducted for injections. If the dosage of excipients and adjuvants exceeds the commonly used range, there may be certain safety concerns, corresponding toxicological studies or relevant literature data shall be provided to prove that the dosage is safe.
11. Nonclinical and/or clinical bridging studies shall be conducted when data from the pharmaceutical comparability study are insufficient to support a change in comparability. In case of application for exemption, there shall be sufficient reasons and basis.
12. Specify the selection, source, grade, specification and test method information of excipients. The excipients shall generally be of medicinal origin and comply with pharmaceutical standards and “Quality Control of Raw Materials and Excipients for the Production of Biological Products” in Chinese Pharmacopoeia.
13. Modification of information on process control parameters for critical manufacturing process steps and intermediate products. The comparison of the manufacturing process control before and after the change is made.
14. Specify whether there is any change in raw materials and excipients (manufacturer, grade, test method, specification), equipment, drug product process (process method, parameters, filling method/volume, etc.) and scale (batch size), specification (test items, standard limit and analytical method), vaccine packaging materials and containers for production. If involved, provide the supporting basis.
15. If involved, describe in detail the change of in-process control parameters and ranges as well as the basis. If applicable, detail any new analytical methods and conduct methodological validation.
16. In case of any change in the adsorption or formulation sequence of the combined vaccine, the compatibility of each component of the vaccine should be mainly investigated, and the quality comparability study and immunogenicity comparison study of the drug product before and after the change should be carried out.
17. If involved, provide information on adventitious agent assessment.
18. If involved, provide information on the quality of raw materials used for the manufacture of adjuvants before and after the change.
19. If involved, the flow chart of adjuvant manufacturing process before and after the change shall be provided, and the key steps of manufacturing process, key process parameters and in-process control strategy shall be specified.
20. If involved, at least three consecutive batches of commercial scale adjuvant manufacturing process validation should be conducted, and the quality comparison study of the adjuvant before and after the change and the stability comparison study of the adjuvant before and after the change should be conducted.
21. If involved, revise the adjuvant specifications.
22. Perform manufacturing process verification of at least one batch of vaccine at commercial production scale (e.g., the batch scale covers routine production, the production process meets the predetermined in-process control standards, the product meets the specification, etc.), and compare the in-process control and batch analysis data before and after the change.
23. Demonstrate that the in-process control parameters do not have an impact on the critical quality attributes of the vaccine.
24. If involved, revise relevant contents of package insert and package label.
25. Explain the reason for the change. Prepare the manufacturing process flow chart, indicate the process steps, in-process control parameters and raw and auxiliary materials used, and show the material addition link. Briefly describe the manufacturing process.
26. If applicable, propose diluent specifications.
27. Perform batch release testing of three consecutive batches of diluent after change, and perform comparability analysis with historical data before change.
28. Diluent stability and product reconstitution stability studies with new diluent are performed and stability comparability is analyzed with the historical pre-change data.
29. The diluent for reconstituted lyophilized products should comply with the provisions of the Pharmacopoeia. If it is not included in the Pharmacopoeia, the process and standards should be fully demonstrated.
30. If involved, update the information of the equipment, and provide the comparison information of the similarities and differences of the operating principles and key technical parameters of the new equipment and the replaced equipment. Perform validation or revalidation on the equipment. If involved, provide the leaching/extraction study data of equipment.
9. Other Changes (Finished Product)
Prerequisites:
① The equipment used before and after the change is unchanged or equivalent.
② The change in manufacturing process and/or in-process controls is due only to the change in batch size.
③ It does not involve safety and quality issues, and the change is not related to recurring deviations or stability concerns in production.
④ The sterility level of the vaccine has not changed.
⑤ The change of equipment will not affect other equipment. The equipment before and after the change is based on the same operating principle, or the material in contact with the vaccine of the equipment before and after the change does not change.
⑥ The same validated manufacturing process is implemented before and after the change. The sterility levels / microbial limit levels of the drug product are not affected.
⑦ Equivalent equipment (with similar equipment design, same operating principle, same or higher grade product contact materials. Equivalent equipment shall provide product of the same quality as the product processed by the previous equipment) is used to replace the current equipment.
⑧ The delivery device is not in direct contact with the vaccine, and only the durability and precision of the delivery device are required.
⑨ The stability of the vaccine should not be reduced as a prerequisite for the modification of storage conditions.
⑩ The vaccine manufacturing process and production quality control method, formulation, specification, packaging materials and containers in direct contact with drugs, storage conditions and other aspects have not changed, and the stability test was conducted according to the stability test protocol reported at the time of marketing registration of the vaccine.
⑪ Changes in storage conditions after reconstitution/dilution of the finished product, etc.
⑫ The shorter shelf life of the vaccine and/or tighter storage conditions of the vaccine are not associated with recurrent deviations in manufacturing or stability concerns; There are no major safety concerns.
⑬ Extension of the shelf life based on the results obtained from an approved stability study protocol using at least three batches of commercial-scale drug products, including extension of the commercial shelf life on the packaging of the vaccine, the time allowed for use after opening, dilution or reconstitution, etc. Extension of the shelf life of the vaccine due to changes in the manufacturing process or existing pharmaceutical excipients in the formulation does not fall within the scope of such changes; Shelf life changes that change the approved stability protocol due to changes in testing methods are also not covered by such changes.
Technical requirements:
1. Explain the reason for change and describe the specific items of change. Provide the necessity, scientificity and rationality of the change.
2. If involved, provide the name (full name), address (specific to plant/workshop, production line) and responsibilities of the production and inspection plants involved in the change; as well as the working principle, production capacity, manufacturer and model of the changed production facilities and equipment. Carry out the validation of the changed production facilities and equipment. Pay attention to the compatibility of the performance, working principle and production capacity, manufacturer and model of the production facilities and equipment with the manufacturing process before and after changes.
3. Carry out manufacturing process validation of at least three consecutive batches of commercial scale products. It should be clarified whether the scale of the validation batch is consistent with the designed manufacturing capacity and an analysis that is representative of the manufacturing process should be performed (e.g., whether the range of routine manufacturing scale can be covered; whether worst-case process conditions can be represented). Validation should include the analysis of consecutive production batches meeting their predefined in-process controls and specifications. If involved, carry out validation of filling and lyophilization process, aseptic process validation, etc.
4. If applicable, provide the comparability study protocol for changes, and carry out the comparability studies on process and quality before and after changes.
5. Provide results under accelerated and/or forced degradation conditions for at least 3 months (or until nonconformance) of the pre- and post-change commercial scale drug product, unless otherwise required. Provide at least 3-6 months (or until nonconformance) of real-time/actual stability data for pre- and post-change commercial scale drug product. Comparability studies are performed for pre- and post-change drug product stability under accelerated and/or forced degradation and real-time/actual conditions. Pre-change data can be historical stability testing results. For multi-dose products, a commitment should be made to provide in-use stability data at the end of the shelf life of the post-change drug product to demonstrate consistent quality of the post-change product over the actual multiple use period. If involved, transport stability studies should be performed.
6. Develop the stability study protocol of the vaccine, commit to continue the long-term stability study to confirm the full shelf life of the vaccine under normal storage conditions, and commit to report any nonconformance in the long-term stability study.
7. If necessary, special safety test (e.g., local irritation test) shall be conducted for injection.
8. Nonclinical and/or clinical bridging studies shall be conducted when data from the pharmaceutical comparability study are insufficient to support a change in comparability. In case of application for exemption, there shall be sufficient reasons and basis.
9. If involved, update the information of the equipment, and provide the comparison information of the similarities and differences of the operating principles and key technical parameters of the new equipment and the replaced equipment. Perform validation or revalidation on the equipment. If involved, provide the leaching/extraction study data of equipment.
10. If involved, provide process flow description, and specify the information of control strategy of critical manufacturing process steps and intermediate products. The formulation process description shall reflect the concept of “point formulation”. Provide the comparative information of process parameters and control measures before and after change.
11. If involved, the feasibility of shared equipment in contact with the product should be verified and specific procedures for cross-switching should be described.
12. Explain the reason for change. Describe the selection basis for packaging materials and containers, and provide information on rationality and suitability. If involved, provide the information on bundling review of vaccine packaging materials and containers. The pharmaceutical packaging materials banned or eliminated by the state shall not be used.
13. Compare the information on vaccine packaging materials and containers before and after the change, such as description, composition materials, size, structural composition, specification, test method and batch analysis data.
14. Provide validation data of new packaging process. Compare the batch analysis data of three consecutive batches of products produced using new packaging with approved packaging materials and containers. If necessary, an extended quality study should be conducted to demonstrate that the quality of the vaccine in the packaging materials and containers before and after the change is comparable.
15. According to domestic and foreign guidelines, the closure integrity and compatibility of the new packaging materials and containers should be studied. For compatibility study, it is feasible to refer to relevant guidelines at home and abroad. In principle, the quality and stability of the vaccine should not be reduced after changing the packaging materials and containers.
16. If applicable, make appropriate revision on the contents, such as specifications, package inserts and package label sample.
17. If applicable, specify the batch formulation. The batch formulation should include a list of all ingredients used in the manufacturing process of the vaccine, the amount of each ingredient in each batch, including overages, and the specifications for each ingredient.
18. Detail the delivery device information. For the change of delivery system device, the corresponding study shall be conducted according to the characteristics of delivery device to prove that the accuracy of administration dose is consistent before and after the change.
19. Clarify the proposed shelf life and/or storage conditions. If applicable, provide approved stability testing protocol and commitment. Vaccines should be stored in accordance with the pharmacopoeia and other relevant provisions.
20. If there is change in stability study protocol, provide an updated post-approval stability study protocol and commitment, along with a justification for developing the protocol.
21. The stability test results covering the proposed shelf life under real-time/actual conditions are generally obtained from at least three consecutive batches of vaccines manufactured by the commercial scale process according to the stability test protocol approved at the time of marketing registration of the vaccine. The conditions for stability study should be representative of the actual worst-case conditions, and the packaging methods and materials of the samples for stability study should be the same or similar to those of the marketed vaccines.
22. The shelf life and/or storage conditions of the vaccine should be determined based on the results of long-term stability testing after the change, and extrapolation of the results is not applicable for the vaccine.
23. The comparability study on the stability of vaccine under two storage conditions before and after the change should be conducted. The study items should be comprehensive, and sensitive stability indicators such as potency, high-molecular polymer, degradation products and sterility should be paid attention to. If involved, provide data on stability studies of commercial-scale drug products covering the proposed storage period following changes in storage conditions after reconstitution/dilution. The conditions for stability study should be representative of the actual worst-case conditions, and the containers/packaging materials for stability study should be representative of the actual conditions.
24. Perform manufacturing process verification of at least one batch of drug product at commercial production scale, including the following: the batch scale covers routine vaccine production, the vaccine production process meets predetermined in-process control standards, and the product meets the specification, and compare the in-process control and batch analysis data.
25. If involved, revise the specification. If a new analytical method is used, methodological validation data should be provided to prove that the proposed analytical method is equivalent to or better than the approved analytical method.
VI. References
1. Technical Guidelines for the Management of Changes in the Manufacturing Process of Biological Products. CFDA. 2005.
2. Technical Guidelines for Preclinical Studies of Preventive Vaccines. CFDA. 2005.
3. Technical Guidelines for Quality Comparability Study of Changes in Vaccine Production Site. CFDA. 2014.
4. Technical Guidelines for Clinical Comparability Exercise of Preventive Vaccines. NMPA. 2019.
5. Technical Guidelines for Studies of Pharmaceutical Changes of Marketed Biological Products (Interim). 2021.
6. Technical Guidelines for Aluminum-Adjuvant-containing Preventive Vaccines. NMPA. 2019.
7. Pharmacopoeia of the People's Republic of China (2020)
8. Vaccines, 7th Edition. 2018.
9. Guidelines on procedures and data requirements for changes to approved vaccines.WHO.2014.
10. Guidance for Industry. Chemistry, Manufacturing, and Controls Changes to an Approved Application: Certain Biological Products. FDA. 2021.
11. Guideline on the details of the various categories of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products.EC (2010/C 17/01).
12. Guidelines on the details of the various categories of variations, on the operation of the procedures laid down in Chapters II, IIa, III and IV of Commission Regulation (EC) No 1234/2008 of 24 November 2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products and on the documentation to be submitted pursuant to those procedures (2013/C 223/01).
13. ICH Q5C: Stability Testing of Biotechnological Biological Products.1995.
14. ICH Q5A (R2): Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin.2022.
15. ICH Q5E: Comparability of Biotechnological/Biological Products Subject to Change in Their Manufacturing Process. November 2004.
16. ICH Q12: Technical and regulatory considerations for pharmaceutical product lifecycle management.2019.
VII. Explanation of Terms
Antigen: Any substance capable of activating and inducing an immune response, which are generally recognized and bound by the specific antigen receptors (TCR or BCR) on the surface of T and B lymphocytes, activate T and B cells to proliferate and differentiate, produce immune response effector products (specific lymphocytes or antibodies), bind to effector products, and then exert adaptive immune response effects.
Post Approval Changes: Any changes, such as changes in the manufacturing process, analytical methods (including specifications), storage conditions, etc., that require reporting after obtaining marketing approval and approved changes.
Related Changes: They refer to other changes that accompany or result from a change.
Comparability Study: It refers to a series of activities such as study design, study implementation and data evaluation to analyze and confirm whether the quality of the product before and after the change is equivalent or highly similar, and the existing knowledge is sufficient to predict that the difference in quality attributes will not adversely affect the safety and efficacy of the product.
Comparability Bridging Study: Data from drug product manufactured with the current process is allowed to be extrapolated to drug product manufactured with the changed process by providing data from nonclinical or clinical studies.
In-process Control: A test performed during production to monitor or adjust the manufacturing process to ensure that the intermediates or final products meet the appropriate specifications. Control of the manufacturing environment or equipment is also considered part of the in-process controls.
VIII. List of Abbreviations