News
News
 Position :
Position :
I. Overall requirements
Where a biological product for prevention manufactured overseas and already marketed in China is transferred to be manufactured in China, the domestic applicant should submit a marketing authorization application according to the biological products for prevention of Class 3.3. In principle, the Marketing Authorization Holder (MAH) and manufacturers before and after transfer should be under control by the unified quality management system. The applicant shall first conduct sufficient assessment, if clinical trial could be waived based on the assessment, the applicant could submit the marketing authorization application directly; if the clinical bridging study is needed, the applicant shall submit the clinical trial application and is encouraged to strengthen the communication with drug review authorities.
The applicant should sort out and prepare the application dossier in accordance with the format number and item sequence of M4: Common Technical Document (CTD) for the Registration of Pharmaceuticals for Human Use (hereinafter referred to as CTD). In the special declaration items section of the application form, "application for the transfer of drugs manufactured overseas and already marketed in China to be manufactured in China" should be specified, and the license number of the drug should be indicated.
II. Overall consideration (I) Simplification principles of application dossier
Biological product for prevention referred to in this application dossier requirements mean the vaccine used for human immunization for the purpose of preventing and controlling the occurrence and prevalence of diseases.
Where a vaccine manufactured overseas and already marketed in China is transferred to be manufactured in China, the applicant could submit the original registration application dossier of the vaccine manufactured overseas and the relevant study data of the transfer for domestic manufacturing to support its marketing authorization application. The applicant may sort out the original registration application dossiers in CTD format according to previous application dossiers after marketing authorization approval of the vaccine, and in combination with the actual situation of the transfer for domestic manufacturing, prepare the data of CMC study and comparability study that are carried out in China. Combined with risk assessment and the studies on the change, the submission of relevant non-clinical and/or clinical study data can be simplified or exempted.
For the dossier which are simplified or exempted for submission, the relevant item number and name should be retained, noted with "no relevant study " or "not applicable" under the item, and the reasons should be stated.
(II) Change risk assessment
According to the changes of vaccines transferred to domestic manufacturing, the applicant should carry out risk assessment in accordance with the principle of case-by-case analysis with reference to ICH Q9 - Q12 and other relevant guidelines, and combined with prior knowledge, process development data, production platform data, previous manufacturing experience of similar varieties, manufacturing experience with relevant production lines and GMP compliance, and conduct a comprehensive assessment of the cumulative effect that may be caused by the associated changes. Risk factors to be considered include, but are not limited to:
1. Marketing and manufacturing maturity or degree of technical control: clinical studies during vaccine development and post-marketing use experience before the transfer, whether the domestic applicant has manufacturing experience of same varieties, degree of control and differences of quality management system, whether the overseas MAH has experience in transferring to other global manufacturing sites, etc.
2. Manufacturing process: Whether complicated manufacturing process steps are involved, whether the products whose stability is susceptible to transport are involved, etc.
3. Characteristics and complexity of the product itself: Whether novel adjuvants, adjuvant systems or complex preparation system of special delivery system are involved.
4. Risk of associated changes: Whether other significant changes that affect product quality attributes are involved, etc.
The technical requirements and application dossier requirements vary with change risk assessment results. The applicant should make a complete study design and study plan based on risk assessment. The applicant should reasonably judge and determine the risk degree of the proposed changes according to the results of risk assessment, so as to make the related management and execution of change strategy, change plan/protocol, studies on the change and change implementation more scientific, compliant and effective.
In principle, other than a change in manufacturing sites, other changes that adversely affect the quality attributes of vaccine products should be avoided or minimized as far as possible. It is recommended that the bacterial (viral) strains for production, cell matrix for production, formulation of the drug product, raw materials and excipients used for production, adjuvants, diluents, and packaging materials in direct contact with drugs should be consistent with those before transfer. The manufacturing process should be consistent with that before the transfer as far as possible.
For the transfer of drug substance and complex drug product, further non-clinical and/or clinical bridging study should be considered to assess and ensure that the quality, safety and efficacy of the product after change is not reduced. In the case of application for simplification or exemption of relevant studies, the applicant should provide the justifications.
(III) Associated changes
When it is transferred for domestic manufacturing in China, it is usually accompanied by associated changes. For associated changes that are unavoidable during the transfer process, the applicant should provide the rationale and necessity for the change as well as an assessment of the potential impact on the vaccine.
Where multiple changes with lower risk are associated, the risk of overall change may increase. Therefore, it is recommended to pay attention to the cumulated influence of multiple associated changes on vaccine safety, effectiveness, and quality controllability. Comprehensive comparability study data of the product before and after transfer should be submitted according to the requirements of ICH Q5E: Comparability of Biotechnological/ Biological Products Subject to Changes in Their Manufacturing Process, the Technical Guidelines for Quality Comparability Study for the Change of Vaccine Manufacturing Sites, the Technical Guidelines for CMC Change Study of Marketed Biological Products (Interim), Technical Guidelines for CMC Change Study of Marketed Vaccines (Interim) and other relevant change guidelines. Non-clinical and/or clinical study data should be provided if necessary.
(IV) Communication and exchanges
The applicant is encouraged to communicate with the relevant drug regulatory authorities and technical organizations in accordance with the relevant requirements of the Provisions for Post-approval Changes of Drugs (Interim) on key nodes such as the expected change situation, comparability study protocol and content, and tests and inspections required, so as to reasonably design the study protocol for the transfer and speed up and standardize the transfer of related products.
III. Requirements for application dossier (I) Module 1
The application dossier should be prepared according to M4 Module 1: Administrative Information and Prescribing Information. Based on simplification requirements, the following aspects should also be considered:
1. The certification documents of the marketed drug
The description of previous applications and approval/disapproval documents of the drug manufactured overseas and already marketed in China shall be provided.
2. Certification documents for transfer to domestic manufacturing in China
The notarized documents evidencing the MAH of the vaccine manufactured overseas agrees to the transfer for domestic manufacturing in China should be provided, with the Chinese translation attached.
A statement of the relationship between the overseas MAH and the domestic applicant/manufacturer, a statement that they are controlled by the unified quality management system and related supporting documents should be provided.
3. Package inserts, labels, specifications, and manufacturing and testing procedures
The differences in vaccine’s package inserts, labels, specifications, and manufacturing and testing procedures before and after transfer, as well as the revisions thereof should be provided.
4. Other necessary certification documents
It is recommended to put the above documents into 1.3.9 Other product information related materials under 1.3 Product Information Related Materials in Module 1.
(II) Module 2
The applicant should submit documents in CTD format, and prepare and organize the application dossier according to the actual situation of the transfer of overseas manufacturing to domestic manufacturing in China. The information required and special considerations are as follows:
For the quality overview, the applicant should provide a summary tabulation/chart of the overall comparison before and after the transfer, and provide the main results and conclusions of the comparison studies before and after the transfer under the relevant items. If there are associated changes other than a manufacturing site change, the studies information shall be summarized under the corresponding item, including but not limited to basis of the changes, assessment of the changes and relevant comparability studies.
Non-clinical overview, clinical overview and summary can be prepared according to the content of studies in Modules 4 and 5.
(III) Module 3
The applicant should submit documents in CTD format, and prepare and organize the application dossier according to the actual changes of the vaccines subject to the transfer of overseas manufacturing to domestic manufacturing in China. The information required and special considerations are as follows:
1. Bacterial (viral) strains for production and cell matrix for production
The bacterial (viral) strains for production and cell matrix for production should be consistent before and after transfer.
If seeds for production are directly supplied by the Transferor, the Transferor shall provide the corresponding study data, including the control measures of the corresponding Quality Management System (QMS) and transportation stability studies.
Where the seed bank is re-established in China after the transfer, the bacteria (virus) seed bank and cell bank after the transfer should be the same as the primary and/or master bank of the overseas products before the transfer, and comprehensive testing and comparability study data of the seed bank after the transfer should be provided.
The preparation and quality control of bacteria (virus) seed bank should meet the requirements of the Chinese Pharmacopoeia or overall not lower than those for the vaccine before transfer. Any discrepancy should be explained and provided with supporting information.
If the master cell bank/seed bank is re-established in China, the applicant should provide the verification and testing report of the master cell bank/master seed bank issued by the NIFDC or third-party test institutes recognized by relevant drug regulatory authorities.
2. Raw materials and excipients for production
The raw materials used for production and excipients should be consistent before and after transfer.
Where the raw materials and excipients are not changed, the original registration application dossier can be used for the dossier of quality studies of the raw materials and excipients, such as, 3.2.P.4.3 Validation of Analytical methods, 3.2.P.4.4 Justification of Specifications, and other items.
If there is any change to the manufacturer of raw materials and excipients, validation and comparability studies should be carried out with reference to the Technical Guidelines for CMC Change Studies of Marketed Biological Products (Interim) and other guidelines. The data of comparison with the raw materials and excipients used by the transferor should be provided, and the consistency of important physicochemical indicators and specifications should be explained. The change risk analysis report of raw materials should be provided clearly in the corresponding sections of 3.2.S.2.6, 3.2.P.2.3 Manufacturing Process Development and 3.2.R Comparability Study.
For cases such as changes in the manufacturer of key excipients for special preparations, in addition to CMC comparability studies, the necessary non-clinical and/or clinical bridging study data should be provided
3. Adjuvants
Adjuvants and adjuvant systems should be consistent before and after transfer.
Where there is no change to the adjuvant or adjuvant system and only transportation is involved, the original registration application dossier of vaccine manufactured overseas can be used, and the transportation stability data and etc. should be provided.
If adjuvants and adjuvant systems are transferred to be manufactured in China or there are other changes, a comparability study before and after transfer should be conducted with reference to the relevant guidelines for post-marketing changes and adjuvant related guidelines, including comparative studies of different stages of adjuvants and vaccine preparations, and necessary non-clinical and/or clinical bridging study data should be provided.
The corresponding study data should be put into the excipient section of 3.2.P.3 and 3.2.A.3.
4. Study of manufacturing process and formulation of drug product
The formulation of drug product should be consistent before and after transfer.
The detailed comparison information of main production equipments, manufacturing process, process parameters, production process control and testing methods, and production scale with those before transfer should be provided and explained in combination with the diagrams/tables.
The detailed information of manufacturing and testing facility, production equipments, formulation development and composition, production scale, steps and parameters of manufacturing process, intermediate control items and limits, packaging materials, suppliers of raw materials and excipients after transfer should be provided. In principle, the validation study data of at least three consecutive commercial batches, and comparative analysis data of process control capabilities before and after transfer, as well as comparative analysis data with historical data before transfer should be provided.
Where there is no change to the formulation and process, production equipment, or packaging materials in direct contact with the drug (e.g., microneedles and atomization, etc.), the original registration application dossier of vaccine manufactured overseas can be used for product development related items, such as 3.2.P.2.2.1 Formulation Development, 3.2.P.2.3 Manufacturing Process Development, etc.
5. Characterization
Quality characteristics of vaccines should be consistent before and after transfer, and supporting data should be provided.
The historical batch results before transfer, and comparability analysis data before and after transfer should be provided, to demonstrate that the transfer causes no changes in quality attributes of vaccines.
The characterization study data of expansion of multiple batches of samples after transfer should be provided, including structural identification, physicochemical properties, vaccine efficacy and impurity levels, and the batch-to-batch consistency should be analyzed.
6. Quality control
In principle, the process control and specifications (including testing items, specification limits and analytical methods) of the vaccines before and after transfer should meet the requirements of the Chinese Pharmacopoeia, and overall not be lower than those for the vaccines before transfer. Any discrepancy should be explained and provided with supporting information.
The study data on the control product/reference product should be provided, including comparative study data with the control product/reference product before transfer, and the difference between the old and new control product/reference product.
In case of the transfer of a testing site or other specification changes involved, the technical data should be provided to support the transfer of methodology.
7. Packaging system
In principle, the packaging materials and containers in direct contact with drugs and drug delivery devices should be consistent. The comparative data on packaging materials and containers in direct contact with drugs as well as drug delivery devices before and after transfer should be provided.
Where there is no change to packaging materials and containers in direct contact with the drug and delivery devices, in combination with the changes in manufacturing process, the original registration dossier on the compatibility of the packaging system and other studies data on the changes should be provided. If there is any change to the supplier, the sufficient basis for the change and relevant study data should be provided.
8. Stability study
The stability study data of at least three batches of vaccine intermediates (if involved), drug substance and drug product after transfer should be provided, and the stability comparability study with historical batches of vaccines before transfer should be conducted. The proposed shelf-life of vaccines and supporting analysis data thereof should be provided.
(IV) Module 4
The non-clinical bridging studies data on comparing the products before and after transfer should be provided with reference to the guidelines for post-marketing change of vaccines, such as the Technical Guidelines for Quality Comparability Study for the Change of Vaccine Manufacturing Site.
Local irritation tests and allergy tests are usually performed for safety consideration (except for oral vaccines), and animal immunogenicity studies may be considered for effectiveness. If the quality comparative results are not accepted, or if there is a large difference in the products before and after the change, or the comparative study items are not sufficient, more safety and/or efficacy studies, such as repeat-dose toxicity study, may be conducted.
(V) Module 5
On the basis of sufficient demonstration that the sample quality before and after transfer is comparable and the necessary non-clinical bridging studies meet the relevant requirements, clinical trials are generally not required and clinical study data can be exempted from submission. Where it cannot demonstrate that the samples are comparable before and after transfer after sufficient CMC comparative study and/or non-clinical bridging study, further consideration can be given to applying for a clinical trial, and submitting relevant application dossiers as required.
IV. Relevant requirements
(I) Regarding the requirements for number of transfers and multiple strengths
A drug manufactured overseas cannot be transferred multiple times to different domestic MAHs for manufacturing in China via this pathway. For instance, once the overseas MAH A has transferred a drug to the domestic Applicant B, the MAH A may not transfer this drug to other domestic applicants during the period when Applicant B holds a valid drug registration number for this drug.
Where there are multiple strengths for one single drug product, some of these strengths may be transferred. If the domestic applicant intends to add other untransferred strengths, the Applicant shall submit corresponding applications in accordance with the present Dossier Requirements for Application. Different strengths of the same drug shall be transferred to the same domestic applicant.
(Ⅱ). Inspection and testing
Inspection for registration and registration tests should be initiated based on risks according to the Provisions for Drug Registration and the Working Procedure for the Initiation of Inspection and Tests for Drug Registration (Interim).
According to the degree of risk of the specification changes, if there is no impact on the specification (including testing items, specification limits and analytical methods), the specification verification may be exempted, and the full-item test should be conducted for the transferred vaccines. If there is any change in testing items, analytical methods and/or specification limits, upon sufficient risk assessment, the specification verification may be conducted on the specific items, and a full-item test may be conducted for the transferred vaccines.
Under the circumstance that the moderate change in manufacturing site specified in the Technical Guidelines for CMC Change Study of Marketed Biological Products (Interim) after transfer is met, the registration test can be exempted, but full-item test of the first three batches of drug products marketed should be conducted according to the Measures for the Administration of Lot Release of Biological Products.
(III) GMP Compliance Inspection
Any transfer to a domestic drug MAH and a domestic manufacturer shall be subject to GMP compliance inspection. During the GMP compliance inspection, attention should be paid to the dossiers provided by the applicant regarding the overseas MAH and the domestic applicant as well as the domestic manufacturer being controlled by a unified Quality Management System (QMS).
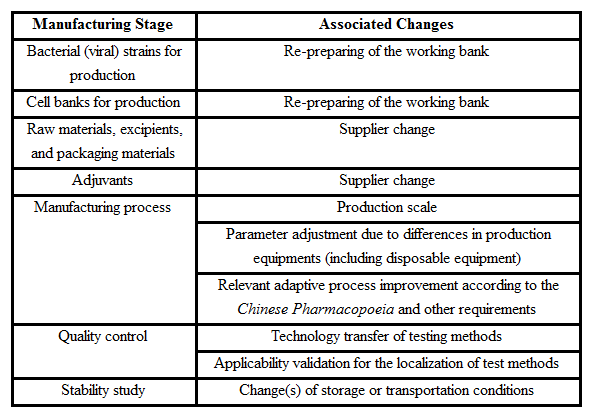
Annex Examples of Common Associated Changes during Transfer