

 ホーム
ホーム 最新情報
最新情報 機構紹介
機構紹介 国際交流
国際交流 サイトについて
サイトについて一、「医療機器登録管理弁法」と「体外診断試薬登録管理弁法」の修訂に関する全体構想と原則とは何か?
「医療機器監督管理条例」における原則と要求に従い、「弁法」を修訂するものである。修訂の全体構想と「医療機器監督管理条例」修訂の全体構想の整合性を保ち、分類管理を基礎とし、リスクの程度を根拠としながら、医療機器登録と届出に関する具体的な要求を確定する。医療機器の登録は行政許可制度の一項目であり、また食品医薬品監督管理機関が医療機器登録申請者の申請に対し、法定手順に従い、市販予定の医療機器の安全性、有効性の研究およびその結果を全体的に評価し、その申請を許可するかどうかを決める過程である。医療機器の届出とは、医療機器届出人が食品医薬品監督管理機関に届出の資料を提出し、食品医薬品監督管理機関が検査に備えてそれらの資料を保管することである。第一類医療機器の届出は、製品のリスクを考慮に入れたうえで設けられた行政監督管理手段である。届出人は行政機関に資料を提出し、行政機関はそれらの資料に対する「形式審査」を行い、届出人に届出受領書を発行し、届出済の情報を発表する。届出と資料保管を通して情報を収集し、後続的な監督と検査を行う。法規の要求に適わない場合、企業が速やかに是正するよう促すか行政処罰などの行政行為を行わなければならない。
二、医療機器届出と登録はそれぞれどの機関で申し込めばよいか?
第一類医療機器の場合は届出管理を実施する。第二類、第三類医療機器の場合は登録管理を実施する。国産第一類医療機器の届出は、届出人が現地の区レベル食品医薬品監督管理機関に関係資料を提出する必要がある。国産第二類医療機器の場合は省、自治区、直轄市の食品医薬品監督管理機関が審査を行い、許可後に医療機器登録証を発行する。国産第三類医療機器の場合は国家食品医薬品監督管理総局が審査を行い、許可後に医療機器登録証を発行する。輸入第一類医療機器の届出の場合、届出人は国家食品医薬品監督管理総局に関係資料を提出する必要がある。輸入第二類、第三類医療機器の場合、国家食品医薬品監督管理総局が審査を行い、許可後に医療機器登録証を発行する。香港、マカオ、台湾地区の医療機器の登録と届出は輸入医療機器の規定に参考にして行う。
三、医療機器届出に必要な提出資料とは?
第一類医療機器の届出に必要な提出資料は安全性・リスク分析報告書、製品技術要求、製品検査報告書、臨床評価資料、製品取扱説明書および最小販売包装ラベルの設計仕様書、生産製造情報、証明文書、適合性に関する声明などである。
四、医療機器登録に必要な提出資料とは?
医療機器登録に必要な提出資料は申請表、証明文書、医療機器安全性・有効性関係基本要求事項リスト、総括資料、研究資料、生産製造情報、臨床評価資料、製品リスク分析資料、製品技術要求、製品登録検査報告書、取扱説明書とラベル仕様書、適合性に関する声明などである。
体外診断試薬登録に必要な提出資料は申請表、証明文書、総括資料、主要原材料の研究資料、主要生産技術および反応体制の研究資料、性能分析評価資料、陽性判定値または参考区間確定資料、安定性研究資料、生産および自主検査の記録、臨床評価資料、製品リスク分析資料、製品技術要求、製品登録検査報告書、製品取扱説明書とラベル仕様書、適合性に関する声明などである。
五、医療機器関係臨床試験の実施について、どのような規定があるか?
医療機器関係臨床試験は、医療機器関係臨床試験品質管理規範の要求に従い、関係資格を取得した臨床試験機構で行われなければならない。臨床試験サンプルの生産は医療機器品質管理体制の関係要求に適わなければならない。第三類医療機器の臨床試験が人体に高いリスクを与える恐れがある場合、、国家食品医薬品監督管理総局の許可のもと行われなければならない。第三類医療機器の目録は国家食品医薬品監督管理総局が制定、修正して発表することになる。臨床試験の審査許可とは国家食品医薬品監督管理総局が申請者の申請に対し、臨床試験実施予定医療機器のリスク、臨床試験方案、臨床におけるメリットとリスクの比較分析などについて総合的な分析を行い、臨床試験の実施を許可するかどうかを決めるプロセスである。医療機器の臨床試験は許可後3年以内に実施されなければならない。期限切れになっても実施されなかった場合、元の許可書類が自動的に廃止する。臨床試験の実施が必要であるなら、再度申請しなければならない。
六、医療機器登録関係審査許可の期限は?
登録申請を受理した食品医薬品監督管理機関は受理後の3勤務日以内に申請資料を技術審査機構に転送しなければならない。技術審査機構は60勤務日以内に第二類医療機器登録の技術審査評価を完了させ、90勤務日以内に第三類医療機器登録の技術審査評価を完了させなければならない。外部専門家を招聘しての審査もしくは薬品と医療機器を組み合わせた製品に関する医薬品審査機構の合同審査を行う必要がある場合、技術審査機構は所要時間を書面で申請者に告知しなければならない。技術審査プロセスで申請者による追加資料提出が必要である場合、技術審査機構は一回で追加資料のすべての内容を通達なければならない。申請者は1年以内に通達に従いすべての追加資料をまとめて提出しなければならない。技術審査機構は追加資料受領後の60勤務日以内に技術審査を完了させなければならない。ただし申請者が追加資料を提出する時間は審査期限に算入しないものとする。登録申請を受理した食品医薬品監督管理機関は技術審査完了後の20勤務日以内に許可するかどうか決定しなければならない。安全性、有効性要求に適う場合、登録を許可し、許可決定後の10勤務日以内に医療機器登録証を発行し、審査済みの製品技術要求仕様書を添付文書として申請者に提出しなければならない。
七、医療機器登録証のフォーマットは?
付表を参考のこと。
中華人民共和国医療機器登録証(フォーマット)
登録証番号:

許可機関: 許可日: 年 月 日
有効期限: 年 月 日
(許可機関印)
中華人民共和国医療機器登録証(体外診断試薬用フォーマット)
登録証番号:
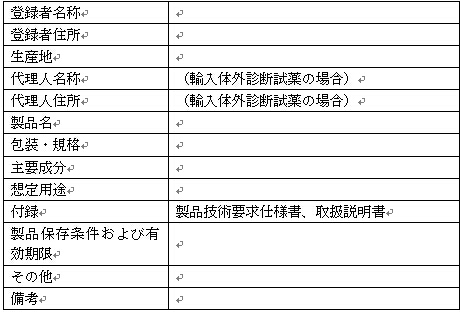
許可機関: 許可日: 年 月 日
有効期限: 年 月 日
(許可機関印)
八、医療機器類製品技術要求とは何か?どのように作成するか?
製品技術要求は主に医療機器完成品の性能指標と検査方法を含む。そのうち、性能指標とは客観的に判定できる完成品の機能、安全性指標および品質制御に関するその他指標を指す。
医療機器類製品技術要求の作成は国の関係法律・法規に従って行われる必要がある。医療機器製品技術要求は規範的で通用する専門用語を使わなければならない。特別な専門用語を使う場合、明確に定義をつけなければならない。医療機器類製品技術要求における検査方法の各項目内容に番号をつける場合、原則として性能指標の各項目内容の番号に合わせてつけなければならない。医療機器類製品技術要求における文字、数字、公式、単位、記号、図表なども標準化要求に適わなければならない。例えば、医療機器類製品技術要求の内容が国家標準、業界標準または中国薬局方を引用する場合、引用した内容の有効性を保証し、関係標準の番号、時間および中国薬局方のバージョンを明記しなければならない。
(出所:CFDAサイト2015-02-05)