

 ホーム
ホーム 最新情報
最新情報 機構紹介
機構紹介 国際交流
国際交流 サイトについて
サイトについて国家食品医薬品監督管理総局は、医薬品臨床試験に関する研究を促進し、申請者と研究者が医薬品開発における全体的な戦略と単独的な臨床試験計画を制定する際に技術的指導を行い、同時に医薬品技術評価に参照を提供するために、「医薬品臨床試験の一般考慮指導原則」を制定し、2017年1月20日に発表した。当該指導原則は発表日から施行開始になる。その全文は以下のとおりである。
医薬品臨床試験の一般考慮指導原則
一、概要
医薬品臨床試験の一般考慮指導原則(以下「指導原則」と略称。)は、現在、国家食品医薬品監督管理総局が医薬品臨床試験の研究を行う際の一般考慮事項である。本指導原則を制定するのは申請者と研究者が医薬品開発における全体的な戦略と単独的な臨床試験計画を制定する際に技術的指導を行い、同時に医薬品技術評価に参照を提供するためである。なお、上市後の医薬品の適応症を増やす際に、臨床試験を行う場合も本指導を参照できる。本指導原則は主に化学医薬品と治療用バイオ製品に適用する。
二、臨床試験の基本原則
(一)被験者保護
1、関係法律と法規の実施
医薬品臨床試験はヘルシンキ宣言を遵守し、国家食品医薬品監督管理総局が制定した「医薬品臨床試験品質管理規範」などの関係法律と法規に従って実施しなければならない。
2、安全性基礎の備え
あらゆる医薬品臨床試験を行う前に、関係非臨床研究または以前の臨床研究の結果が当該薬物の推薦されたヒトでの試験研究において試験を行う場合、受けられる安全性の基礎を十分に説明できなければならない。
医薬品研究開発のプロセスで、薬理学と毒性学の専門家及び臨床関係専門家などは薬理学的、毒性学的データと臨床試験データをダイナミックに評価し、臨床試験が被験者に与えられる安全性のリスクを評価すべきである。必要に応じて実施中または実施予定の臨床試験プロトコールを調整しなければならない。
医薬品臨床試験に参与する各側は自らの職責を果たし、被験者保護の責任を負わなければならない。
(二)臨床試験の基本方法
1、臨床試験の一般規律
医薬品研究開発の本質は医薬品有効性と安全性の問題を提起し、研究を通して解答することである。臨床試験とは、ヒトでの試験を通し、医薬品を使用して疾病の予防、治療、診断を行う際の特定問題を解答することである。通常は以下のとおりに2種類の方法で臨床試験を記述する。
試験の段階によって、Ⅰ期臨床試験、Ⅱ期臨床試験、Ⅲ期臨床試験、Ⅳ期臨床試験と分類する。
研究の目的によって、薬理学的臨床研究、模索的臨床試験、確証的臨床試験、上市後における研究と分類する。
上記2通りの分類方法はいずれも制限があるが、相互補完でダイナミックで実用的価値を持つ臨床試験ネットワークを立ち上げた(図1を参照に)。
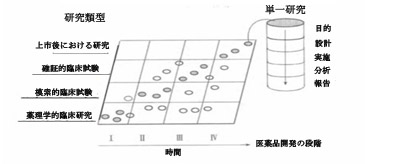
図1、臨床研究段階と研究類型の関係
(中実円はある段階で通常行われる研究類型を表す。中空円は通常ではないが、行われる可能性のある研究類型を表す。)
概念実証(Proof of Concept,POC)とは医薬品の薬理学的効果が臨床的価値に転化できると検証することで、普通、早期臨床研究の段階で行われ、安全用量で使用される医薬品の有効性の信号を探る手段で、医薬品開発リスクを削減できる。
本指導原則は研究目的分類を主な主軸とし、臨床試験を記述することとする。
臨床薬理学研究の目的は耐性について評価し、薬物動態学的及び薬効学的特徴を究明したうえで関係記述を行い、薬物代謝と薬物相互作用を探り、薬物活性を分析することにある。
模索的臨床試験の研究目的はターゲット適応症の継続研究で投薬プランを練り、有効性と安全性に関する確証的研究の設計、エンドポイントの設定、研究方法の選定などに根拠を提供することにある。
確証的臨床試験の目的は有効性と安全性を確認し、医薬品登録のサポートとして、ベネフィット/リスク関係評価の根拠を提供し、且つ用量と効果の関係を確定することにある。
上市後における研究の目的は薬物が一般患者、特別患者及び/又は環境におけるベネフィット/リスク関係の認識をを再認識し、稀な副作用を発見し、投薬プランの整備に臨床的根拠を提供することにある。
2、目標をガイドとする臨床研究
薬物臨床研究開発の戦略として、目標をガイドとする臨床試験研究開発モデルを採用しなければならない。臨床研究全体計画は明確な終極的目標と明晰な研究ルートを設定しなければならない。各臨床試験はいずれも明確な試験目的を持たなければならない。
3、臨床試験の各段階における戦略決定
臨床試験のプロセスは、絶えずに戦略を決定するプロセスである。毎回、臨床試験終了後、関係階段のベネフィット/リスクの評価を行い、研究開発を中止するか、または継続するかを決定すべきである。データがリスクを明らかに存在すると示した場合(有効性が欠け、もしくは安全性に問題がある)、なるべく早く臨床試験を中止しなければならない。データが研究開発の将来性を示した場合、既存の研究データを踏まえたうえで、臨床試験を次第に推し進めなければならない。臨床研究計画も研究結果によって、適当に調整しなければならない。例えば、臨床試験の有効性検証結果はより多くのヒト試験を通して薬理学的研究を行う必要があるという提示することもできる。場合によって、臨床試験の選別結果に基づき、確定した適応症を変更または除去しなければならない。
4、臨床試験プロセスの規範化
臨床試験を行う際に、科学的に設計、実施と分析を行い、プロセスの規範化と結果の信憑性を保障し、報告書を漏れなく、如実に記入しなければならない。
5、安全性に関する全体的考慮事項
通常、臨床試験のサンプル量は有効性を考慮に入れた決定したもので、安全性評価にとって足りない可能性がある。安全性評価は十分なサンプル量と暴露時間が必要である。重篤ではない患者が長期的に医薬品を使用する際の安全性を評価する場合、普通は以下の原則に従わなければならない。
薬物研究開発の段階において、定性的、定量的に医薬品の安全性関係特徴に記述しなければならない。臨床試験では、安全性の観察期間は医薬品を長期的に使用予定期間と同様にするよう推奨する。医薬品の潜在的な危険を十分探究するために、サンプル量の設定にあたって以下の事項を考えなければならない。
(1)薬物の暴露時限。
(2)暴露時限中における有害事象の発生時間と重篤度。
(3)有害事象は治療時間の延長によって変化の趨勢。
通常、重長期的に医薬品を使用する重篤ではない患者を研究する場合、暴露のよくある有害事象を発覚することに必要なサンプル量は約1500例である(短期間暴露を含む)。初の有害事象は普通最初の数ヶ月に発生する。例えば、臨床治療期間が6ヶ月間を例とし、300-600例のサンプルで通常に発生する用害事情の発生率(例えば、全体発生率は0.5%-5%)と変化趨勢(増加又は減少)を暴露する。治療時間の延長に伴い、一部の有害事象は発生頻度も重篤度も上昇する。また、一部の有害事象は6ヶ月間後に現われるため、当該有害事象を発覚するために、少なくとも、100名の患者が12ヵ月月以上の暴露が必要がある。
状況によっては、実情に基づいてサンプル量を増やし(減らし)、もしくは観察周期を延長する必要もある。
三、臨床研究計画における方法学的注目すべき事項
申請者は臨床試験を行う前に、臨床研究開発の全体計画を制定しなければならない。研究開発計画は主に2つの方面の内容を含む。一つ目は、非臨床研究の中で、充分なデータを持っているかどうかを注目すること。それによって、臨床試験設計の中で被験者の安全性と有効性を指導し、臨床試験で使用する薬物は安定している品質基礎をもっているかどうかに用いる。二目は目標をガイドとする臨床試験全体設計の考え方の下で、いかに各段階と各目的の臨床試験を設計するかを注目すること。
(一)臨床試験を行うための基礎
1、非臨床研究
非臨床研究の設計は今後の臨床試験にかかわる要素を考慮に入れなければならない。それらの要素とは、(1)各被験者の中で暴露する医薬品の総量及び持続する時間、(2)薬物の特徴(例えば、半減期が長いこと)、(3)ターゲットとする適応症、(4)特別患者への投与(例えば、出産適齢期の女性)、(5)投薬ルートである。
既存の毒性学的、薬理学的、薬物動態学的な非臨床研究の結果などをまとめ、非臨床研究データが実施予定の臨床試験をどこまでサポートできるかを判断する(詳細は関係指導原則を参照)。
(1)安全性研究
ヒトを対象とする初の臨床試験を行う前に、薬物動態学的、薬理学的、毒性学的な非臨床研究データを総合的に評価しなければならない。非臨床研究データはヒトへの初投薬時の用量と安全な暴露時間を確定するに十分な情報を提供すると同時に、薬物研究に関する薬理学的、毒性学的情報も提供しなければならない。
(2)薬理学的及び薬物動態学的研究
非臨床研究データは臨床試験を行う基礎であると同時に、臨床試験の研究方向にも影響を与える。臨床試験を行う前に、申請者は普通、以下の非臨床研究の情報を取得しなければならない。
薬物の主な役割を果たす薬理学的根拠(薬物の役割発揮メカニズム。バイオマーカーなど分子標的研究を含む。)用量効果又は濃度効果の関係及び作用持続時間実施可能な投薬ルート一般薬理学、薬物が主要器官系に果たす薬理学的作用と生理学的効果⑤医薬品の吸収、分布、代謝と排出に関する研究結果
⑥場合によっては、医薬品のゲノムやプロテオームなどに関する研究結果が必要になる。
2、薬物品質に関する研究
臨床試験に用いる処方の特徴を十分述べ、あらゆる入手可能なバイオアベイラビリティ関係データを提供しなければならない。処方は薬物開発の段階に適応しなければならない。提供した製剤は一定用量範囲内の一連の研究に適当するものなら、理想的である。薬物研究開発期間中、同一薬物の異なる処方を使用する可能性があるため、生物学的同等性またはそのたの方法で製剤の関連を考慮し、開発全体計画で臨床研究の結果を理解するのは非常に重要である。
(二)臨床試験研究開発のプロセス
1、臨床薬理学的研究
新薬上市申請は薬物の安全性と有効性に関する評価をサポートする臨床薬理学的研究がある必要がある。研究内容は主に薬物がヒトに与える効果(治療効果と副作用)、ヒトにおける薬物への対処(薬物動態学関係)、薬物の代謝及び物質バランス、用量?暴露量?効果関係、薬物相互作用、薬物ゲノム関係データ、定量薬理学的研究、特別患者向け臨床薬理学的研究、患者グループ向け薬物動態学的研究などを含む。臨床試験の段階によって、臨床薬理学的研究の任務と内容も異なる。
臨床薬理学的研究は、通常、臨床試験の早期段階で行うが、薬物開発の必要で別の段階で行うことも可能である。臨床薬理学的研究は普通、治療目的ではないため、健康な医学ボランティアを対象に行い、疾病そのものが研究結果に与える影響を抑える。ただし、一部の薬物、例えば、細胞毒性薬は、健康な人間に有害であるため、患者を対象に研究を行うしかない。
臨床薬理的研究は通常、ランダム化、盲検法、対照試験の形をとるが、場合によっては、その他の設計も可能である。
(1)耐性試験
ヒトを対象とする耐性試験は、ヒトが耐えられる最大用量を確定し、ヒトで最初に現われる副作用の特質も発見できある。投薬方法は単回投与と複数回投与がある。
ヒトを対象とする耐性試験を行う前に、2通りの情報を把握しなければならない。一つは非臨床研究の評価結論である。もう一つは関係薬物または類似薬物に関する既存の臨床研究又は文献を研究すること。それらの情報はヒト試験の安全用量を事前算出し、臨床副作用モニタリング指標を選定するには重要な意味を持つ。
①ヒト試験初回投与安全用量の確定
耐性テスト関係臨床試験における薬物初回投与安全用量の確定は関係指導原則と方法を参照するように(定量薬理学的研究の方法など)。
ヒトで初回の臨床試験を行う際の最大推奨初回投与量(Maximum Recommended Starting Dose,MRSD)は副作用発生しないと想定する投与量である。初回投与で副作用の発生を防止すると同時に、適切な用量を選定し、合理的な速度と進行度で耐性試験関係臨床試験の最終的目標(例えば、耐性関係、薬効学的または薬物動態学的指標で判断する)を達成しなければならない。
②耐性試験の終止に関する考慮事項
ヒトを対象とする耐性試験を行う前に、試験終止基準を定めなければならない。具体的には、どのような有害事象または暴露濃度で、投与量逓増試験を終止すべきである。
終止基準を定める際には、以下のような事項ついて注目する必要がある。健康な医学ボランティアを対象に試験を行う際に、なるべく被験者に健康被害を与えないこととする。薬物の想定する適応症にかかった患者グループの特徴に基づいて終止基準を確定する。また、潜在的リスクの高い薬物の場合、動物試験で獲得した安全性データとヒト安全性との種間差が存在するかどうかに注意しなければならない。バイオ製剤及び新しいメカニズム、新しい標的、新しいルートなどで開発された薬物の場合は特に注意しなければならない。
③単回投与と多回投与の耐性研究
薬物を初めてヒトに使用する際、まずMRSDを計算、確定し、それからMRSDで1回投与し、耐性試験を行う。潜在的リスクの高い薬物の場合、参考できる安全性データが限られることや、動物試験の結果とヒトとの相違の存在可能性などを考慮に入れ、初のヒト耐性試験は少数個体を対象に行うべきである。例えば、大分子バイオ薬物の場合、初のヒト耐性試験はリスクを低減し、被験者を保護するために、一人一人の被験者を対象に行い、安全性データを獲得したうえで試験を進めることとする。試験実施機構は試験に適当する施設、設備と職員を備えなければならない。
通常、多回投与耐性試験は単回投与耐性試験の結果が出た後に、且つ単回投与のヒト薬物動態学的試験の結果が出た後に行われる。この一回耐性試験と一回薬物動態的試験の結果は多回投与耐性試験の設計を指導できるわけである。具体的には、用量と投与ルートの選定、投与と食事の関係、有害事象の特質と程度などである。
ほとんどの場合には、単回投与耐性試験と単回投与薬物動態学的研究、単回投与耐性試験と単回投与薬物動態学的研究はいずれも同時実施が可能である。耐性試験は早期臨床安全性試験であるため、より信憑性の高い結果を出すために、条件が備えれば、ランダム化、二重盲検、プラシーボ効果で試験を設計することを推奨する。
(2)薬物動態学的研究
ヒトにおける薬物吸収、分布、代謝と排出の特徴に関する研究は普通、開発計画全体を貫く。薬物動態学的特徴を確定するのは臨床薬理学的研究の重要目標の一つである。薬物動態学的研究は関係指導原則を参照できる。
薬物動態学的研究は多数の独立研究を通じて評価を行うことも、薬効学的研究、安全性と耐性研究の一部として評価を行うこともできる。そして、薬物暴露、分布、排除率の分析、プロトタイプ薬物または蓄積可能な代謝物、潜在的な薬物間相互作用の予測などに重要な意味を持つ。
単回投与の薬物動態学的研究は、ヒトが薬物を吸収する速度と程度、投薬量と薬物濃度の関係、薬物の半減期などを把握するためである。単回投与の研究結果を得た後、多回投与を研究を行う。重複投薬後の薬物の吸収程度、濃度が安定値に達するまでの時間、体内の薬物蓄積程度などがどうなるのかを探究する。通常、単回投与の研究で究明した薬物半減期の結果は、多回投与の研究に投与時間間隔の設定に重要な参考となる。例えば、半減期が短い薬物の場合、多回投与の研究で、普通、24時間内に何度も投与する必要があり、さらに作用機序などのデータに基づいて総合的に分析、判断しなければならない。
薬物用量と濃度の関係を把握するために、少なくとも低、中、高という3レベルの用量でそれぞれ単回投与と多回投与の薬物動態学的研究を行う必要がある。また、用量はMRSDと最大耐性量の間に設定しなければならない。
内服薬の場合、通常、食べ物がバイオアベイラビリティに与える影響を研究すべきである。その研究は放出の形が変更する可能性のある薬物にとって重要である。また、普通、単回投与の研究で、適当に用量を設定し、食べ物が薬物に与えるを影響を探究しなければならない。
特別グループの薬物動態学的情報が研究の範囲にも考慮に入れるべきである。例えば、臓器機能障害(腎臓または肝臓疾患)の患者、高齢者、児童、妊娠中または哺乳中の女性及び人種別グループなどに関する研究。
今後は専門性の高い問題を解答するために、各グループ向け薬物動態学的研究はもちろん、各種類の薬物動態学的研究が必要になる。
(3)薬効学的評価
上述したとおり、開発中薬物の特徴に基づき、薬効学的研究と血中薬物濃度研究は健康なボランティアまたは患者を対象に行うことが可能である。適当な測定方法があれば、薬効学的データに基づいて患者向け試験で薬物の活性と潜在的な有効性を早期評価ができ、且つその後の研究で、適応症患者への投与量と投与ルートの確定に根拠を提供できる。
2、模索的臨床試験
患者を対象に初めて有効性の模索という目的で臨床試験を行うのは、模索的臨床試験の始まりと思われる。
模索的臨床試験は通常、被験者グループの同質性を確保し、被験者を厳密にモニタリングするために、被験者を厳格に選定する必要がある。
早期模索的臨床試験は多数の研究設計を取られる。並行対照法と自身対照法を含む。その後の臨床試験は通常、ランダム化と対照研究になる。
模索的臨床試験の重要目標の一つは、確証的臨床試験に薬物投与量と投与プランを確定することである。早期模索的臨床試験は通常、投与量逓増の形をとり、薬物の用量と効果の関係に対する初歩的な評価を行う。後期模索的臨床試験は、検討対象となる適応症に対し、通常並行群間比較を行い、用量?効果の関係を探究する形をとる。模索的臨床試験の投薬量は通常、薬理的臨床研究で提示した最大耐性量より小さく設定する。最大耐性量を超えた場合、適当に薬理学的臨床試験を追加で行い、サポートとなるデータを提供しなければならない。
模索的臨床試験のその他の目的は次の臨床試験で設定するエンドポイント、治療プラン(合併投与を含む。)とターゲット患者グループ(例えば、軽度と重度疾患の比較)について評価することである。その目的はサブグループのデータと多数のエンドポイントの分析で果たすことができ、分析の結果は次の段階における模索的臨床試験または確証的臨床試験に使用できる。
3、確証的臨床試験
治療ベネフィットの獲得を重要な目的とする。
確証的臨床試験は模索的臨床試験で獲得した薬物有効性と安全性に関す初歩的証拠をより一層確証し、上市許可取得にあたって十分な証拠を提供するためでもある。その研究内容は用量?効果関係に対するさらなる模索、またはより幅広い患者、疾病の異なる段階、合併投与に関する研究になる。長期服用予定薬物の場合、暴露延長試験は模索的臨床試験の段階で始まったにもかかわらず、通常、確証的臨床試験で行う。長期投与薬物と高齢者向け薬物の臨床安全性データに関する考慮事項については、本指導原則は記述しないこととする。確証的臨床試験は薬物添付文書の完璧整備に重要な臨床情報を提供しなければならない。確証的臨床試験で、グループ間薬物動態学的研究、薬物ゲノム研究などを同時に行うことができる。
4、上市後における研究
研究目的によって、医薬品上市後における研究は以下の2種類に分けられる。
(1)監督管理部門の要求に従って行うもの。あらゆる関係法規の要求に準じて行う上市後の研究で、行わなければならない医薬品上市後安全性研究と登録許可書類における完成しなければならない研究内容を含む。
(2)自主実施のもの。監督管理部門の要求で、申請者または第三者の承諾又は自ら行う研究である。上市後における研究は通常、付加した薬物間相互作用、長期投与または大規模サンプル使用時の安全性、薬物経済学及び関係薬物使用による許容範囲内適応症の臨床的エンドポイントをサポートする研究(例えば、死亡率/発病率研究など)などを含む。
研究の目的と内容によって、研究モデルまたはツールを適当に選択して研究を展開しなければならない。研究方法は薬理学的臨床研究、臨床試験、観察による薬剤疫学研究、メタアナリシスなどである。研究方法によって、研究結果も異なり、解決できる問題も違う。
5、補充申請事項
最初の医薬品上市許可を取得した後、関係法律や法規などに基づいて新しい適応症と適応症変更の研究、新しい投薬プランと投与ルートまたはそのた患者群に関する研究を行うことが可能である。新規投与量と新規処方または合併投与の研究を行う場合、薬理学的臨床研究も追加で行わなければならない。元の研究計画または上市後における研究と応用に関するデータを使用するのは、一部の研究を省略する可能性がある。
(三)特別に考慮に入れる事項
1、薬物代謝産物の研究
主な活性的代謝産物を鑑別し、関係薬物動態学的研究を行う。
2、薬物相互作用の研究
潜在的な薬物間相互作用があった場合、臨床研究の早期段階で関係研究を行うことを推奨する。薬物併合使用をよく行う場合は、非臨床研究またはヒト試験(可能であれば)で関係薬物相互作用を研究する必要がある。これは、自身以外の薬物の吸収または代謝を変更する薬物、または自身の動態がほかの薬物に影響される薬物にとって特に重要である。
3、特別グループ
一般グループと比べて、特別グループはベネフィット/リスク比が異なり、もしくは投与量と投与時間を調整する必要があるかもしれない。そこで、特別グループを対象とする特別な臨床試験を行うべきである。腎臓、肝臓機能不全患者に関する薬物動態学的研究は発生可能な薬物代謝または排出変更の評価に特に重要な意味を持つ(関係指導原則を参照)。
(1)妊娠中女性に関する研究
研究対象となる薬物は妊娠期間に使用する予定がなければ、妊娠中女性は研究範囲外としなければならない。患者が臨床試験期間中に妊娠した場合、通常は試験を中止し、倫理委員会に届け出、妊娠期間、胎児と児童の状況をフォローして評価しなければならない。妊娠期間中に使用する予定のある薬物臨床研究が妊娠中女性と関わる場合にも、妊娠期間、胎児と児童の状況をフォローして強化するのが非常に重要である。
(2)哺乳期間中女性に関する研究
可能であれば、母乳における薬物またはその代謝産物の含有量を測定する必要がある。哺乳期間中女性を臨床試験の被験者にした場合、薬物が乳児に与える影響についてモニタリングし、必要に応じてその子供のフォローする必要がある。
(3)児童に関する研究
児童に関する研究の内容は、薬物の既存認識、及び成人とその他年齢別児童グループの外挿可能性によって決定される。一部の薬物は早期研究開発段階で児童に向けになるかもしれない。
児童が使用するように期待される薬物の場合、年齢適当な児童グループで研究を展開し、評価を行うべきである。臨床試験が児童にかかわる場合、年上グループから年下グループへという順番で研究を展開すべきである。
4、薬物ゲノム学的研究
薬物ゲノム学的研究は非臨床研究の段階で開始してもいい。早期臨床試験で、ゲノムに関する投与量、有効性または安全性データはサンプル量に制限され、確定的ではないが、後期臨床試験(確証的臨床試験)における投与量設定と被験者選定に根拠を提供できる。研究戦略から言えば、早期臨床試験で正確で有効的な投与量を採用し、患者サブグループを合理的に選定し、敏感で正確な薬物動態学的なバイオマーカーもしくは合理的なエンドポイントを利用すれば、研究者は早く薬物の標的直接調整作用を把握でき、有効性などの情報を獲得でき、機序または概念を検証する目的を果たす。さらに、開発戦略と方向性の決定に臨床的根拠を提供できる。
四、単数の独臨床試験の考慮事項
臨床試験の目的、設計、実施、結果分析及び報告書作成にあたって、以下の原則に従わなければならない。研究を行う前に、研究の各部分を臨床試験プロトコールに記入しなければならない。
(一)目的
臨床試験の目的をはっきり述べなければならない。臨床試験の目的は薬物動態学的バロメーターへの評価でも、薬物の薬理的、生理的効果への評価でも、薬物の有効性または安全性への模索、確証でもよい。
(二)設計
合理的な臨床試験設計は価値のある結論を獲得する前提である。臨床試験設計は並行対照、群逐次、交差、階乗、適応性などがあるが、並行対照の採用を推奨する。臨床試験の目的を果たすために、申請者は被験者グループの特徴をはっきり述べ、対照物を合理的に選択し、主要と副次的なエンドポイントを記述し、サンプル量試算の根拠を提供しなければならない。臨床症状、徴候と実験室検査指標に基づいて安全性を評価する方法についても述べなければならない。また、予定より早く試験を終止した場合、被験者フォローはいかに行うのかも説明しなければならない。統計分析に関する設計は関係指導原則を参照する。
1、被験者グループの選定
被験者グループを選択する際に、研究の段階と適応症及び既存の非臨床と臨床研究のデータを考慮に入れなければならない。早期試験では、被験者のグループ変化ついては厳しい選別基準によって大体同質的な被験者を選出できるが、試験を進めるにつれて、ターゲット患者における治療効果を反映させるために、被験者の幅を拡大しなければならない。
研究開発の進捗状況と安全性への注目度によって、一部の試験は厳密なモニタリングのもとで行わなければならない(例えば、入院など)。
極めて特別な事情があった場合は除外だが、通常は被験者は同時に2つ又はそれ以上の臨床試験に参加しなければならない。安全性を確保し、延滞の影響を防止するために、十分な時間的間隔がなければ、被験者は重複して臨床試験に参加してはいけない。
出産適齢期の女性が臨床試験に参加する際には、通常、避妊措置をとらなければならない。
男性医学ボランティアの場合、試験で薬物暴露がその性的パートナーまたは子供に被害を与えるかどうかを考えなければならない。被害があれば(例えば、試験で変異誘発効果があり、もしくは生殖器系に毒性がある薬物にかかわる場合)、試験で適当に避妊措置を提供しなければならない。
2、対照グループの選定
臨床試験は合理的な対照を選択しなければならない。対照の類型はプラセボ対照、陽性対照、自身対照、薬物用量間対照、無治療対照、歴史対照などになる。対照類型の選択は試験の目的によって決め、倫理的な問題をコントロール可能にすると同時に、科学関係要求に準じなければならない。通常はプラセボ対照法の採用を推奨するが、それ以外の対照法を選択する場合には、事前に意思疎通を行わなければならない。歴史(外部)対照は論証後、特別な状況のもとで、採用してよいが、推論の間違いでリスクを高めることに特に注意しなければならない。
陽性対象法は試験で使用する薬物を慎重に選択しなければならない。適当な陽性対照は(1)公認され、幅広く使用されもの(2)根拠に基づく医療に関する証拠をもっていること(3)有効性が再現できるもの。試験設計を行う際に、関係臨床使用の進捗状況を考慮に入れるべきである。
3、サンプル量の試算
試験規模は研究対象となる疾病、研究目的とエンドポイントに影響される。サンプル量は治療効果の予測、異変程度の予測、統計分析方法、偽陽性率、偽陰性率などで確定すべきである。また、場合によっては、薬物の安全性を確認するには、膨大なデータが必要になる。
4、研究指標
研究指標については明確に定義しなければならない。定義とは、指標の属性(定性、定量、半定量)及び具体的な観察方法である。
試験エンドポイントは薬物動態学的バロメーター、薬効学的測定値、薬物有効性と安全性など薬物作用に関する研究指標の評価で使用される。主要エンドポイントは主な臨床効果を反映し、研究の主要目的によって選択しなければならない。副次的なエンドポイントはその他の薬物作用の評価で使用され、主要エンドポイントと関連しても関連しなくてもいい。エンドポイント及びその分析については、研究計画で予め述べなければならない。
代替エンドポイントは臨床エンドポイントに関連する指標であるが、臨床価値を裏付ける直接証拠ではない。代替エンドポイントで臨床エンドポイントを予測できる場合、代替エンドポイントは主要指標になる。
臨床エンドポイントを評価する方法は、主観的でも客観的でも、その正確さ及び応答性(時間の変化に対する敏感度)が公認されたものでなければならない。
5、偏差制御方法
(1)ランダム化
対照試験で、ランダムにグループを分けるのは被験者グループ間比較可能性の確保と選択による偏差の低減を優先するためである。ランダム化は通常、グループ分けランダム化と分層ランダム化の形をとる。多数の分層要素を考えなければならない場合、ダイナミックな方法でランダムに被験者を分け、各層の均衡性を保つ必要がある。
(2)盲検法
盲検法は二重盲検法、単盲検法、開放的盲検法に分けられる。盲検法は研究結果の偏差を制御するための重要な手段である。二重盲検法とは、被験者、研究者、臨床関係申請者がいずれも被験者グループ分けを知らない試験法である。単盲検法とは、被験者がグループ分けを知らない試験法である。二重盲検試験でプラセボ対照を行う場合は、通常、単一のシミュレーション技術を使用して盲検状態を維持する。陽性薬物対照を行う場合、陽性薬物と試験対象の薬物は感覚で分別でき、もしくは投与ルートが異なるなら、二重シミュレーション技術を使用して盲検状態を維持する必要がある。シミュレーションが難しいなら、その他の遮蔽措置で二重盲検を行うことが可能だが、試験計画で関係技術マニュアルを明記しなければならない。盲検をどこまで実施するかを問わず、データ管理スタッフと統計分析スタッフはいずれもブラインド状態でなければならない。通常、二重盲検法の採用を推奨する。
(3)依存性
試験での被験者薬物使用状況は試験プランに明記し、具体的な使用状況は記録して保存しなければならない。
(三)実施
薬物開発は本指導原則に基づいて実施しなければならない。研究者は試験計画に従って作業しなければならない。開発計画を修正する必要があれば、添付ファイルを提供し、修正の理由と合理性を説明し、倫理委員会に報告して許可を取得しなければならない。開発中は有害事象の情報を集め、記録しなければならない。また、関係監管機関に速やかに安全性データについて報告しなければならない。
(四)分析
臨床試験計画は専門的な統計分析プランを含み、試験目的と設計に適当するものでなければならない。
統計分析プランは被験者グループの分け方、効果関係指標設定方法を考慮に入れて作成しなければならない。統計分析はなるべく治療意図の原理による解析(ITT解析)という形で行い、プランに乖離、違反する被験者については分析を行う際に考えなければならない。ランダムにグループを分けた後に被験者を取り除くことはなるべく少なくし、やむを得ず取り除く場合は具体的な原因をあげなければならない。統計方法及び統計分析用ソフトウェアとそのバージョンについて、説明しなければならない。プラン中間分析を行う時間の選定についても説明しなければならない。
臨床試験データの分析は試験計画で予定したとおりに行わなければならず、いずれの偏差と乖離があった場合は説明しなければならない。
一部の試験は前もって終了するのは予定であれば、試験計画で総合Ⅰ類ミス率(偽陽性率)の制御について説明しなければならない。研究プロセスでサンプル量の調整が必要になった場合、その根拠を提供し、ランダム状態で調整することを推奨する。調整後のサンプル量は元の計画より大きく設定しなければならない。調整によって試験の完璧性を損なわないことも説明しなければならない。
臨床試験はいずれも安全性データを収集し、グラフと表で表示し、有害事象の重篤度と研究対象となる薬物との相関性によってそれらのデータを分類、分析しなければならない。
試験データの統計分析プラン、統計分析報告書及び研究報告書の作成はいずれも関係指導原則を参照しなければならない。
(五)報告書
臨床試験報告書は関係指導原則に基づいて作成しなければならない。
(出所:CFDAサイト2017-01-20)